Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (120)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Humidity Calibration of Dynamic Vapor Sorption (DVS) InstrumentДокумент5 страницHumidity Calibration of Dynamic Vapor Sorption (DVS) Instrumentleizar_death64Оценок пока нет
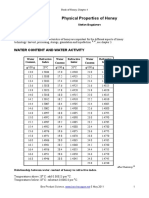
- Bogdanov S. (2016) - Physical PropertiesHoney - Honey Book, Chapter4Документ8 страницBogdanov S. (2016) - Physical PropertiesHoney - Honey Book, Chapter4dangthanhsonОценок пока нет
- PREFORMULATION STUDIEsДокумент8 страницPREFORMULATION STUDIEsTanishaОценок пока нет
- Avitech Technical Bulletin: Influence of Powder Properties On Premix ProductionДокумент6 страницAvitech Technical Bulletin: Influence of Powder Properties On Premix Productionamamùra maamarОценок пока нет
- TDS Tamol NN 9104Документ5 страницTDS Tamol NN 9104UtpalОценок пока нет
- Megger Book The Complete Guide To Electrical Insulation TestingДокумент35 страницMegger Book The Complete Guide To Electrical Insulation TestingTimmyJuriОценок пока нет
- Potassium FeldsparДокумент6 страницPotassium FeldsparSengupta VivekОценок пока нет
- Physical Separation of Components in A Mixture: (Organic and Inorganic Substances)Документ25 страницPhysical Separation of Components in A Mixture: (Organic and Inorganic Substances)Venice Joy CelociaОценок пока нет
- Acids, Bases and SaltsДокумент20 страницAcids, Bases and SaltsTunde DabiriОценок пока нет
- Experiment No. 12: Preparation of Precipitated Sulfur USPДокумент7 страницExperiment No. 12: Preparation of Precipitated Sulfur USPAlessandra Joy M. RoceroОценок пока нет
- 3 2 4 Mahle Fuellmengenhandbuch Agrar enДокумент44 страницы3 2 4 Mahle Fuellmengenhandbuch Agrar enJanos HallaОценок пока нет
- (BS EN 1097-10 - 2002) - Tests For Mechanical and Physical Properties of Aggregates. Determination of Water Suction HeightДокумент14 страниц(BS EN 1097-10 - 2002) - Tests For Mechanical and Physical Properties of Aggregates. Determination of Water Suction HeightAdelОценок пока нет
- US3659785 - Weather Modification Utilizing Microencapsulated MaterialДокумент5 страницUS3659785 - Weather Modification Utilizing Microencapsulated MaterialHerb GreenОценок пока нет
- 8th English Science 2 PDFДокумент160 страниц8th English Science 2 PDFSantosh ParvatikarОценок пока нет
- Hygroscopic Deliquensce Efflorescence PDFДокумент2 страницыHygroscopic Deliquensce Efflorescence PDFJedd MattОценок пока нет
- ASHRAE 2011 Gaseous and Particulate Contamination Guidelines For Data CentersДокумент22 страницыASHRAE 2011 Gaseous and Particulate Contamination Guidelines For Data Centersrc100% (1)
- Preformulation and Product Development: Bishnu Prasad KoiralaДокумент59 страницPreformulation and Product Development: Bishnu Prasad Koiralasaloni patelОценок пока нет
- Powder: Mr. Suraj Mandal M.Pharm PharmaceuticsДокумент37 страницPowder: Mr. Suraj Mandal M.Pharm PharmaceuticsAshwani Guleria100% (1)
- International Standard: Norme InternationaleДокумент11 страницInternational Standard: Norme InternationaleSanthoshОценок пока нет
- Regeneration of Composite Desiccant Dehumidifier by Parabolic Trough Solar Collector - An Experimental InvestigationДокумент9 страницRegeneration of Composite Desiccant Dehumidifier by Parabolic Trough Solar Collector - An Experimental InvestigationAqsal Raja BramasthaОценок пока нет
- Calculation Based On DIN 55474Документ2 страницыCalculation Based On DIN 55474Braulio Martinez100% (3)
- Water2 0Документ23 страницыWater2 0aaravparashar97Оценок пока нет
- Routsis Injection Molding Reference GuideДокумент98 страницRoutsis Injection Molding Reference Guidealfauro100% (1)
- Effervescent PharmaceuticalsДокумент13 страницEffervescent PharmaceuticalsPuzhpop92% (12)
- Code of Practice: Investigation and Control of Dampness in BuildingsДокумент20 страницCode of Practice: Investigation and Control of Dampness in BuildingsJoe GaffneyОценок пока нет
- EP 6, Section 5.11Документ1 страницаEP 6, Section 5.11shanahan86Оценок пока нет
- Amy Fang, "How Bake Time and Ingredient Ratios Affect Cookie Texture"Документ6 страницAmy Fang, "How Bake Time and Ingredient Ratios Affect Cookie Texture"MIT Comparative Media Studies/WritingОценок пока нет
- Absorption Chiller Rev3 (MSB) 20-9Документ96 страницAbsorption Chiller Rev3 (MSB) 20-9norshakeela100% (3)
- Ventilation of Cargo HoldsДокумент3 страницыVentilation of Cargo HoldsHarman SandhuОценок пока нет
- 7 TH STSOДокумент5 страниц7 TH STSOKesanam Sp33% (3)