Вам также может понравиться
- Republic V Drugmaker's Laboratories, IncДокумент17 страницRepublic V Drugmaker's Laboratories, IncSheena Reyes-BellenОценок пока нет
- Republic of The Philippines Rep. by The Bureau of Food and Drugs (BFAD) Now Food and Drugs Administration vs. Drugmaker's Laboratories, Inc. and Terramedic, IncДокумент8 страницRepublic of The Philippines Rep. by The Bureau of Food and Drugs (BFAD) Now Food and Drugs Administration vs. Drugmaker's Laboratories, Inc. and Terramedic, IncIRRAОценок пока нет
- 28 Republic Vs Drugmakers LaboratoriesДокумент3 страницы28 Republic Vs Drugmakers LaboratoriesJoyceОценок пока нет
- Republic Vs Drugmaker - S LaboratiriesДокумент2 страницыRepublic Vs Drugmaker - S LaboratiriesHudson Cee100% (1)
- Republic of The Philippines vs. Drugmakers G.R. No. 190837 March 5, 2014Документ10 страницRepublic of The Philippines vs. Drugmakers G.R. No. 190837 March 5, 2014herbs22225847Оценок пока нет
- Republic Vs Drugmaker's LaboratoriesДокумент3 страницыRepublic Vs Drugmaker's LaboratoriesccvillanuevaОценок пока нет
- Republic v. Drugmaker's Laboratories, Inc.Документ3 страницыRepublic v. Drugmaker's Laboratories, Inc.Cristelle Elaine Collera100% (4)
- Republic Vs Drugmakers LaboratoryДокумент7 страницRepublic Vs Drugmakers LaboratoryKim Laurente-AlibОценок пока нет
- Republic Vs Drugmakers Laboratories IncДокумент9 страницRepublic Vs Drugmakers Laboratories IncManelle Paula GutierrezОценок пока нет
- REPUBLIC v. DRUGMAKER'S LABORATORIES, GR No. 190837, 05 March 2014Документ4 страницыREPUBLIC v. DRUGMAKER'S LABORATORIES, GR No. 190837, 05 March 2014Maizee KeenОценок пока нет
- Republic v. Drugmaker's LaboratoriesДокумент10 страницRepublic v. Drugmaker's LaboratoriesJemuel LadabanОценок пока нет
- 2 (G.R. No. 190837)Документ11 страниц2 (G.R. No. 190837)Jay Mark EscondeОценок пока нет
- BUREAU OF FOOD AND DRUGS Vs DRUGMAKERS LABORATORIESДокумент3 страницыBUREAU OF FOOD AND DRUGS Vs DRUGMAKERS LABORATORIESMarinelMarquezОценок пока нет
- Republic of The Philippines, Represented by The Bureau ofДокумент14 страницRepublic of The Philippines, Represented by The Bureau ofNiñoMaurinОценок пока нет
- Of Tbe Llbilippines Tourt: FffilanilaДокумент10 страницOf Tbe Llbilippines Tourt: Fffilanilaariesha1985Оценок пока нет
- Republic of The Philippines, Represented by The BureauДокумент16 страницRepublic of The Philippines, Represented by The BureauCelestino Law0% (1)
- REPUBLIC OF THE PHILIPPINES vs. DRUGMAKER'S LABORATORIES, INC.Документ1 страницаREPUBLIC OF THE PHILIPPINES vs. DRUGMAKER'S LABORATORIES, INC.AreeОценок пока нет
- Republic vs. Drugmaker's Laboratories, Inc. and Terramedic, Inc., G.R. No. 190837, March 05, 2014Документ2 страницыRepublic vs. Drugmaker's Laboratories, Inc. and Terramedic, Inc., G.R. No. 190837, March 05, 2014Elaine Belle OgayonОценок пока нет
- Gov. Luis Raymund F. Villaflurete, Jr. Vs Hon. Jesse M. RobredoДокумент4 страницыGov. Luis Raymund F. Villaflurete, Jr. Vs Hon. Jesse M. Robredojury jasonОценок пока нет
- 1 9 DigestДокумент13 страниц1 9 DigestMaryland AlajasОценок пока нет
- Admelec Week 3Документ16 страницAdmelec Week 3Roizki Edward MarquezОценок пока нет
- 02 - Republic v. Drugmakers Lab. Inc - BarcelonaДокумент3 страницы02 - Republic v. Drugmakers Lab. Inc - BarcelonaIñigo Mathay RojasОценок пока нет
- Admin Law Case DigestДокумент7 страницAdmin Law Case DigestNel PasaizОценок пока нет
- Custom Search: Today Is Monday, January 29, 2018Документ3 страницыCustom Search: Today Is Monday, January 29, 2018reyna amor condesОценок пока нет
- Pharmaceutical and Health Care Association of The Philippines VsДокумент4 страницыPharmaceutical and Health Care Association of The Philippines VsALEXANDRIA RYLE MUNARОценок пока нет
- Gesmundo CO - Zuneca Parmaceutical V Natrapharm, Sep. 08 2020Документ2 страницыGesmundo CO - Zuneca Parmaceutical V Natrapharm, Sep. 08 2020Vel June De LeonОценок пока нет
- ALFI Vs GarinДокумент12 страницALFI Vs GarinDeborah Grace ApiladoОценок пока нет
- Suboxone Ecf Pacer Uscourts Gov Doc 153112462429Документ38 страницSuboxone Ecf Pacer Uscourts Gov Doc 153112462429James LindonОценок пока нет
- Pharma v. DuqueIII Case DigestДокумент3 страницыPharma v. DuqueIII Case DigestALEXANDRIA RYLE MUNARОценок пока нет
- Republic Vs Drugmaker'S Lab (GR No. 190837 March 5, 2014)Документ17 страницRepublic Vs Drugmaker'S Lab (GR No. 190837 March 5, 2014)Reyvenne GraceОценок пока нет
- Venus Commercial Co. Inc. vs. The Department of Health G.R. No. 240764 November 18 2021Документ26 страницVenus Commercial Co. Inc. vs. The Department of Health G.R. No. 240764 November 18 2021rikki marie pajares100% (1)
- ALFI Vs Hon. Janette GarinДокумент12 страницALFI Vs Hon. Janette GarinJulieОценок пока нет
- Alliance Vs GarinДокумент47 страницAlliance Vs GarinA M I R AОценок пока нет
- Epublic of Tbe Jlbilippines Upreme Tourt: !ooanilaДокумент10 страницEpublic of Tbe Jlbilippines Upreme Tourt: !ooanilaMonocrete Construction Philippines, Inc.Оценок пока нет
- AO 56 Amendment - 5th Revision - 14 OctДокумент13 страницAO 56 Amendment - 5th Revision - 14 OctRod PagdilaoОценок пока нет
- Ra 9502Документ3 страницыRa 9502Nica SolisОценок пока нет
- Upreme !court: 31/epublic of Tbe Btlipptn SДокумент37 страницUpreme !court: 31/epublic of Tbe Btlipptn SLynne EvangelistaОценок пока нет
- Facundo T. Bautista For Petitioners.: Custom SearchДокумент4 страницыFacundo T. Bautista For Petitioners.: Custom SearchRogelio BataclanОценок пока нет
- Alliance For The Family Foundation Vs GarinДокумент20 страницAlliance For The Family Foundation Vs GarinRelmie TaasanОценок пока нет
- Admin Law DigestДокумент19 страницAdmin Law DigestRosalia L. Completano LptОценок пока нет
- ADMIN LAW DIGESTsДокумент19 страницADMIN LAW DIGESTsRosalia L. Completano LptОценок пока нет
- Tutorial 070Документ20 страницTutorial 070Jason HenryОценок пока нет
- Colgate-Palmolive Vs JimenezДокумент2 страницыColgate-Palmolive Vs JimenezJohn Garcia100% (1)
- Generics ActДокумент3 страницыGenerics ActAudette PascualОценок пока нет
- G.R. No. 173034 Pharmaceutical Vs DuqueДокумент2 страницыG.R. No. 173034 Pharmaceutical Vs DuqueApoi Faminiano100% (4)
- Cases AdminДокумент217 страницCases Adminxeileen08Оценок пока нет
- Administrative Law Cases For DigestДокумент228 страницAdministrative Law Cases For Digestanon_746511540Оценок пока нет
- United States v. Algon Chemical Inc., A Corporation, and Edward Latinsky, An Individual, 879 F.2d 1154, 3rd Cir. (1989)Документ18 страницUnited States v. Algon Chemical Inc., A Corporation, and Edward Latinsky, An Individual, 879 F.2d 1154, 3rd Cir. (1989)Scribd Government DocsОценок пока нет
- Mod 2 - Admin CDДокумент12 страницMod 2 - Admin CDclarris harline eginaОценок пока нет
- Republic Act 6675Документ5 страницRepublic Act 6675Vinson PatronОценок пока нет
- Statcon Cases 101722Документ14 страницStatcon Cases 101722Junly Pepito PejanaОценок пока нет
- 22 - Pharmaceutical and Health Care Association of The Philippines Vs Duque IIIДокумент3 страницы22 - Pharmaceutical and Health Care Association of The Philippines Vs Duque IIINicholette Jeanne P. Legaspi100% (1)
- BOEHRINGER INGELHEIM PHARMA GMBH & CO. KG Et Al v. UNITED STATES FOOD AND DRUG ADMINISTRATIONДокумент26 страницBOEHRINGER INGELHEIM PHARMA GMBH & CO. KG Et Al v. UNITED STATES FOOD AND DRUG ADMINISTRATIONjsherkowОценок пока нет
- Alliance For The Family Foundation, Philippines, Inc. (ALFI) Et - Al. vs. Hon. Garin (G.R. Nos. 217872 and 221866, 26 April 2017)Документ2 страницыAlliance For The Family Foundation, Philippines, Inc. (ALFI) Et - Al. vs. Hon. Garin (G.R. Nos. 217872 and 221866, 26 April 2017)Arl GamazonОценок пока нет
- Executive OrderДокумент65 страницExecutive Ordercoral2178Оценок пока нет
- 3 Alliance - For - The - Family - Foundation20220703-11-Dfc9dkДокумент20 страниц3 Alliance - For - The - Family - Foundation20220703-11-Dfc9dkCharisse SarateОценок пока нет
- United States Court of Appeals, Tenth CircuitДокумент10 страницUnited States Court of Appeals, Tenth CircuitScribd Government DocsОценок пока нет
- Del Rosario Vs BengsonДокумент6 страницDel Rosario Vs BengsonTim PuertosОценок пока нет
- Alliance v. GarinДокумент21 страницаAlliance v. GarinPepe UrquiolaОценок пока нет
- Spouses Florendo v. CA PDFДокумент5 страницSpouses Florendo v. CA PDFEszle Ann L. ChuaОценок пока нет
- Tiu v. Platinum PlansДокумент4 страницыTiu v. Platinum PlansEszle Ann L. ChuaОценок пока нет
- Tongoy v. CA PDFДокумент11 страницTongoy v. CA PDFEszle Ann L. ChuaОценок пока нет
- Tong Brothers v. IACДокумент4 страницыTong Brothers v. IACEszle Ann L. ChuaОценок пока нет
- Spouses Aguinaldo v. Torres PDFДокумент7 страницSpouses Aguinaldo v. Torres PDFEszle Ann L. ChuaОценок пока нет
- Weldon Construction v. CAДокумент7 страницWeldon Construction v. CAEszle Ann L. ChuaОценок пока нет
- Velasco v. CA (1973)Документ11 страницVelasco v. CA (1973)Eszle Ann L. ChuaОценок пока нет
- Carbonell v. CAДокумент1 страницаCarbonell v. CAEszle Ann L. ChuaОценок пока нет
- Swire Realty v. YuДокумент6 страницSwire Realty v. YuEszle Ann L. ChuaОценок пока нет
- HSBC v. PauliДокумент1 страницаHSBC v. PauliEszle Ann L. ChuaОценок пока нет
- Cabaliw v. SadorraДокумент1 страницаCabaliw v. SadorraEszle Ann L. ChuaОценок пока нет
- House International Building Tenants Association v. IACДокумент1 страницаHouse International Building Tenants Association v. IACEszle Ann L. ChuaОценок пока нет
- Poole-Blunden v. Union BankДокумент1 страницаPoole-Blunden v. Union BankEszle Ann L. ChuaОценок пока нет
- Babao v. PerezДокумент1 страницаBabao v. PerezEszle Ann L. ChuaОценок пока нет
- Yuvienco v. DacuycuyДокумент1 страницаYuvienco v. DacuycuyEszle Ann L. ChuaОценок пока нет
- Ortega v. LeonardoДокумент1 страницаOrtega v. LeonardoEszle Ann L. ChuaОценок пока нет
- Cabague v. AuxilioДокумент1 страницаCabague v. AuxilioEszle Ann L. ChuaОценок пока нет
- Ker v. Lingad, 38 SCRA 524, April 30, 1971Документ11 страницKer v. Lingad, 38 SCRA 524, April 30, 1971Eszle Ann L. ChuaОценок пока нет
- 234 Supreme Court Reports Annotated: Miguel vs. CatalinoДокумент10 страниц234 Supreme Court Reports Annotated: Miguel vs. CatalinoEszle Ann L. ChuaОценок пока нет
- Strycker's Bay Neighborhood Council, Inc. v. KarlenДокумент1 страницаStrycker's Bay Neighborhood Council, Inc. v. KarlenEszle Ann L. ChuaОценок пока нет
- Buenaseda v. Flavier G.R No. 106719 PDFДокумент16 страницBuenaseda v. Flavier G.R No. 106719 PDFEszle Ann L. ChuaОценок пока нет
- Genuino v. de Lima G.R No. 197930Документ26 страницGenuino v. de Lima G.R No. 197930Eszle Ann L. Chua100% (1)
- Ople v. Torres G.R. No. 127685Документ13 страницOple v. Torres G.R. No. 127685Eszle Ann L. ChuaОценок пока нет
- In Re VsДокумент6 страницIn Re VsEszle Ann L. ChuaОценок пока нет
- Ad. BPPK 16Документ119 страницAd. BPPK 16Sachin BagewadiОценок пока нет
- Huizhe Wu, MD Mingyan Liu, MD Shuang Wang, MD Wanyu Feng, MD, PHD Weifan Yao, Bs Haishan Zhao, Bs and Minjie Wei, MD, PHDДокумент10 страницHuizhe Wu, MD Mingyan Liu, MD Shuang Wang, MD Wanyu Feng, MD, PHD Weifan Yao, Bs Haishan Zhao, Bs and Minjie Wei, MD, PHDDyva VanillaОценок пока нет
- S. No. Division Name Purpose Name Fee Paid As Per GSR 1193Документ9 страницS. No. Division Name Purpose Name Fee Paid As Per GSR 1193VIJAY YADAVОценок пока нет
- 2016 - MUFG Global Generic Pharma Industry ReviewДокумент25 страниц2016 - MUFG Global Generic Pharma Industry ReviewNat BanyatpiyaphodОценок пока нет
- Module 2 - Regulatory Approval Process - Lecture NotesДокумент16 страницModule 2 - Regulatory Approval Process - Lecture NotesyvssmanjunathОценок пока нет
- Bioequivalence of Oxcarbazepine Oral Suspension vs. Film-Coated Tablet in Healthy Chinese Male SubjectsДокумент8 страницBioequivalence of Oxcarbazepine Oral Suspension vs. Film-Coated Tablet in Healthy Chinese Male SubjectsdarismendyОценок пока нет
- 1756 0500 4 344 PDFДокумент8 страниц1756 0500 4 344 PDFAjeet SinghОценок пока нет
- STATISTICAL APPROACHES For Dissoprofile CopmarisionДокумент144 страницыSTATISTICAL APPROACHES For Dissoprofile Copmarisiondrs_mdu48Оценок пока нет
- Aurobindo Ar 2016 17Документ228 страницAurobindo Ar 2016 17Anonymous xfjk1AhSОценок пока нет
- Biopharmaceutics Answer Key-BLUE PACOPДокумент48 страницBiopharmaceutics Answer Key-BLUE PACOPChengD100% (5)
- Comparative Study of Generic Drug Approval Process in Eu, Usa and China - A ReviewДокумент16 страницComparative Study of Generic Drug Approval Process in Eu, Usa and China - A ReviewTuyến Đặng ThịОценок пока нет
- Solubility and PermeabilityДокумент3 страницыSolubility and PermeabilityGNCDWОценок пока нет
- Regulatory Perspectives On Strength-Dependent Dissolution ProfilesДокумент11 страницRegulatory Perspectives On Strength-Dependent Dissolution ProfilesJminsОценок пока нет
- An Abbreviated New Drug Application (ANDA)Документ33 страницыAn Abbreviated New Drug Application (ANDA)Nim DCОценок пока нет
- Colchicine TabletsДокумент16 страницColchicine TabletsSaket SharmaОценок пока нет
- Bioavailability o Fdisperse Dosage FormДокумент94 страницыBioavailability o Fdisperse Dosage Formpharmashri5399Оценок пока нет
- Bioavailabilitas Bioekivalen 1Документ28 страницBioavailabilitas Bioekivalen 1DeliaGvin SimatupangОценок пока нет
- Comparing Generic and Innovator Drugs A Review ofДокумент16 страницComparing Generic and Innovator Drugs A Review ofPrachi PandeyОценок пока нет
- Biowaivers Criteria RequirementsДокумент11 страницBiowaivers Criteria RequirementsSaravanan AnnamalaiОценок пока нет
- Reflection Paper On The Dissolution Specification For Generic Solid Oral Immediate Release Products With Systemic ActionДокумент10 страницReflection Paper On The Dissolution Specification For Generic Solid Oral Immediate Release Products With Systemic ActionAnitha KalyankarОценок пока нет
- USP 1150 PharmaceuticalStability MKT PDFДокумент3 страницыUSP 1150 PharmaceuticalStability MKT PDFMuhammad FadhlurrahmanОценок пока нет
- Disolusi Sildenafil (15 MNT, Min 85%)Документ9 страницDisolusi Sildenafil (15 MNT, Min 85%)Katarina KatarinaОценок пока нет
- Drug Approval System of The Philippines PDFДокумент56 страницDrug Approval System of The Philippines PDFPatrick OribelloОценок пока нет
- Public Assessment Report Scientific DiscussionДокумент19 страницPublic Assessment Report Scientific DiscussionTaki JuveОценок пока нет
- Ich m9 Biopharmaceutics Classification System Based Biowaivers Step 5 - enДокумент18 страницIch m9 Biopharmaceutics Classification System Based Biowaivers Step 5 - enmagugurgelОценок пока нет
- WwwbiracnicinДокумент252 страницыWwwbiracnicinCIO White PapersОценок пока нет
- Jurnal 2 Ts 2Документ8 страницJurnal 2 Ts 2h2blockersОценок пока нет
- Medicinal Products DossierДокумент14 страницMedicinal Products DossierBasiaОценок пока нет
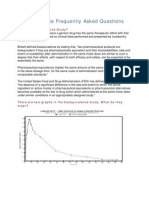
- Bioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Документ4 страницыBioequivalence Frequently Asked Questions: What Is Bioequivalence Study?Ahmed HasanОценок пока нет
- Bioeq Rev-Oxcarbazepine TabsДокумент55 страницBioeq Rev-Oxcarbazepine TabsJagdish ChanderОценок пока нет
- Principles of Welding: Processes, Physics, Chemistry, and MetallurgyОт EverandPrinciples of Welding: Processes, Physics, Chemistry, and MetallurgyРейтинг: 4 из 5 звезд4/5 (1)
- The Aqua Group Guide to Procurement, Tendering and Contract AdministrationОт EverandThe Aqua Group Guide to Procurement, Tendering and Contract AdministrationMark HackettРейтинг: 4 из 5 звезд4/5 (1)
- A Place of My Own: The Architecture of DaydreamsОт EverandA Place of My Own: The Architecture of DaydreamsРейтинг: 4 из 5 звезд4/5 (242)
- Real Life: Construction Management Guide from A-ZОт EverandReal Life: Construction Management Guide from A-ZРейтинг: 4.5 из 5 звезд4.5/5 (4)
- Civil Engineer's Handbook of Professional PracticeОт EverandCivil Engineer's Handbook of Professional PracticeРейтинг: 4.5 из 5 звезд4.5/5 (2)
- Post Weld Heat Treatment PWHT: Standards, Procedures, Applications, and Interview Q&AОт EverandPost Weld Heat Treatment PWHT: Standards, Procedures, Applications, and Interview Q&AОценок пока нет
- Estimating Construction Profitably: Developing a System for Residential EstimatingОт EverandEstimating Construction Profitably: Developing a System for Residential EstimatingОценок пока нет
- Pressure Vessels: Design, Formulas, Codes, and Interview Questions & Answers ExplainedОт EverandPressure Vessels: Design, Formulas, Codes, and Interview Questions & Answers ExplainedРейтинг: 5 из 5 звезд5/5 (1)
- Building Physics -- Heat, Air and Moisture: Fundamentals and Engineering Methods with Examples and ExercisesОт EverandBuilding Physics -- Heat, Air and Moisture: Fundamentals and Engineering Methods with Examples and ExercisesОценок пока нет
- The Complete HVAC BIBLE for Beginners: The Most Practical & Updated Guide to Heating, Ventilation, and Air Conditioning Systems | Installation, Troubleshooting and Repair | Residential & CommercialОт EverandThe Complete HVAC BIBLE for Beginners: The Most Practical & Updated Guide to Heating, Ventilation, and Air Conditioning Systems | Installation, Troubleshooting and Repair | Residential & CommercialОценок пока нет
- How to Estimate with RSMeans Data: Basic Skills for Building ConstructionОт EverandHow to Estimate with RSMeans Data: Basic Skills for Building ConstructionРейтинг: 4.5 из 5 звезд4.5/5 (2)
- 1,001 Questions & Answers for the CWI Exam: Welding Metallurgy and Visual Inspection Study GuideОт Everand1,001 Questions & Answers for the CWI Exam: Welding Metallurgy and Visual Inspection Study GuideРейтинг: 3.5 из 5 звезд3.5/5 (7)
- Sustainable Design and Build: Building, Energy, Roads, Bridges, Water and Sewer SystemsОт EverandSustainable Design and Build: Building, Energy, Roads, Bridges, Water and Sewer SystemsОценок пока нет
- Practical Guides to Testing and Commissioning of Mechanical, Electrical and Plumbing (Mep) InstallationsОт EverandPractical Guides to Testing and Commissioning of Mechanical, Electrical and Plumbing (Mep) InstallationsРейтинг: 3.5 из 5 звезд3.5/5 (3)
- The Complete Guide to Building With Rocks & Stone: Stonework Projects and Techniques Explained SimplyОт EverandThe Complete Guide to Building With Rocks & Stone: Stonework Projects and Techniques Explained SimplyРейтинг: 4 из 5 звезд4/5 (1)
- Building Construction Technology: A Useful Guide - Part 1От EverandBuilding Construction Technology: A Useful Guide - Part 1Рейтинг: 4 из 5 звезд4/5 (3)
- Essential Building Science: Understanding Energy and Moisture in High Performance House DesignОт EverandEssential Building Science: Understanding Energy and Moisture in High Performance House DesignРейтинг: 5 из 5 звезд5/5 (1)
- Fire Protection Engineering in Building DesignОт EverandFire Protection Engineering in Building DesignРейтинг: 4.5 из 5 звезд4.5/5 (5)
- The Complete Guide to Alternative Home Building Materials & Methods: Including Sod, Compressed Earth, Plaster, Straw, Beer Cans, Bottles, Cordwood, and Many Other Low Cost MaterialsОт EverandThe Complete Guide to Alternative Home Building Materials & Methods: Including Sod, Compressed Earth, Plaster, Straw, Beer Cans, Bottles, Cordwood, and Many Other Low Cost MaterialsРейтинг: 4.5 из 5 звезд4.5/5 (6)
- Starting Your Career as a Contractor: How to Build and Run a Construction BusinessОт EverandStarting Your Career as a Contractor: How to Build and Run a Construction BusinessРейтинг: 5 из 5 звезд5/5 (3)
- The Complete Guide to Building Your Own Home and Saving Thousands on Your New HouseОт EverandThe Complete Guide to Building Your Own Home and Saving Thousands on Your New HouseРейтинг: 5 из 5 звезд5/5 (3)
- An Architect's Guide to Construction: Tales from the Trenches Book 1От EverandAn Architect's Guide to Construction: Tales from the Trenches Book 1Оценок пока нет