Вам также может понравиться
- BIO Genetics SplicingДокумент14 страницBIO Genetics SplicingAnonymous SVy8sOsvJDОценок пока нет
- Gene Expression and DNA ChipsДокумент88 страницGene Expression and DNA ChipsAkshitaОценок пока нет
- Gene Expression - Microarrays: Misha KapusheskyДокумент144 страницыGene Expression - Microarrays: Misha Kapusheskymuhammad ichwanОценок пока нет
- BGi RNA-Seq AnalysisДокумент19 страницBGi RNA-Seq Analysisshashikanth_marriОценок пока нет
- Annotating Genomes Using Proteomics Data: Andy Jones Department of Preclinical Veterinary ScienceДокумент22 страницыAnnotating Genomes Using Proteomics Data: Andy Jones Department of Preclinical Veterinary ScienceShaher Bano MirzaОценок пока нет
- Genomes: Number of Base PairsДокумент38 страницGenomes: Number of Base PairsshooberОценок пока нет
- Eukaryotic Gene PredictionДокумент61 страницаEukaryotic Gene Predictionthamizh555Оценок пока нет
- Animal Genomics And: Methods For Genotype DetectionДокумент52 страницыAnimal Genomics And: Methods For Genotype Detectiontoton78tinku85Оценок пока нет
- Metagenomic Shotgun Seq Learning ProgressДокумент19 страницMetagenomic Shotgun Seq Learning ProgressAnisaLFОценок пока нет
- Genome AnnotationДокумент25 страницGenome AnnotationSajjad Hossain ShuvoОценок пока нет
- Gene Prediction-2Документ138 страницGene Prediction-2thamizh555Оценок пока нет
- SAFC Biosciences Scientific Posters - Using Microarray Technology To Select Housekeeping Genes in CHO CellsДокумент1 страницаSAFC Biosciences Scientific Posters - Using Microarray Technology To Select Housekeeping Genes in CHO CellsSAFC-Global100% (1)
- Rasl-Seq Epa Cop 20mar2014Документ19 страницRasl-Seq Epa Cop 20mar2014dupuytrenОценок пока нет
- Agency LawДокумент57 страницAgency LawMisikir Obsa SaanОценок пока нет
- Pant NagarДокумент45 страницPant NagarAshok ChaudharyОценок пока нет
- Science of Living System (BS20001) : - Soumya deДокумент45 страницScience of Living System (BS20001) : - Soumya deMayank PriayadarshiОценок пока нет
- GenomicsДокумент38 страницGenomicsBiplabendu DandapatОценок пока нет
- Biological Sequence Determination: ProteinДокумент68 страницBiological Sequence Determination: Proteinpathcell bioОценок пока нет
- RmzKIPKPEeiixgqCUDoEfA Coursera PlantBioinformatics Lecture02Документ9 страницRmzKIPKPEeiixgqCUDoEfA Coursera PlantBioinformatics Lecture02Ali SherОценок пока нет
- Day 2 General Microarray Lecture - Ver11706Документ48 страницDay 2 General Microarray Lecture - Ver11706amitОценок пока нет
- Aug. 31 - Sept. 4, 2014Документ49 страницAug. 31 - Sept. 4, 2014giapvanthuОценок пока нет
- BS10003 - Transcription and Translation - December 2020Документ38 страницBS10003 - Transcription and Translation - December 2020dhiraj moreОценок пока нет
- Gene Expression RNA SequenceДокумент120 страницGene Expression RNA SequenceMutsawashe MunetsiОценок пока нет
- APPLICATION OF BIOINFORMATICS IN MOLECULAR BIOLOGY AND CURRENT RESEACRH-Dr. Ruchi YadavДокумент105 страницAPPLICATION OF BIOINFORMATICS IN MOLECULAR BIOLOGY AND CURRENT RESEACRH-Dr. Ruchi YadavAlishaОценок пока нет
- Science of Living System: Nihar Ranjan JanaДокумент20 страницScience of Living System: Nihar Ranjan JanaRajnandni SharmaОценок пока нет
- Lecture 21, Protein SynthesisДокумент21 страницаLecture 21, Protein SynthesisNariopolusОценок пока нет
- From RNA-seq Reads To Gene ExpressionДокумент27 страницFrom RNA-seq Reads To Gene ExpressionHoangHaiОценок пока нет
- ExomeДокумент62 страницыExomerajeshbhramaОценок пока нет
- Ritobrata Goswami: Email: Tel: 03222-284570Документ44 страницыRitobrata Goswami: Email: Tel: 03222-284570Ashmit RanjanОценок пока нет
- Prokaryotic Gene ExpressionДокумент28 страницProkaryotic Gene ExpressionNasiha el KarimaОценок пока нет
- Science of Living System: Arindam MondalДокумент48 страницScience of Living System: Arindam MondalSohini RoyОценок пока нет
- Mbt1 B Cloning Expression 14 15Документ91 страницаMbt1 B Cloning Expression 14 15Abhishek SinghОценок пока нет
- HTTP en - Biomarker.com - CN Wp-Content Uploads 2021 04 Nanopore Full-Length RNA-SeqДокумент6 страницHTTP en - Biomarker.com - CN Wp-Content Uploads 2021 04 Nanopore Full-Length RNA-SeqMobio lecturesОценок пока нет
- Online Biological Databases: A/Prof. Ly LeДокумент64 страницыOnline Biological Databases: A/Prof. Ly LeLinhNguyeОценок пока нет
- Data Analysis in Next Generation SequencingДокумент78 страницData Analysis in Next Generation Sequencingparetini01Оценок пока нет
- Nomedatabase (Proteomics)Документ38 страницNomedatabase (Proteomics)Quicker QuickОценок пока нет
- Lecture Notes Algorithms in Bioinformatics I - Prof. Daniel HusonДокумент28 страницLecture Notes Algorithms in Bioinformatics I - Prof. Daniel HusonmsОценок пока нет
- Regulatory Motif Finding: Wenxiu Ma CS374 Presentation 11/03/2005Документ54 страницыRegulatory Motif Finding: Wenxiu Ma CS374 Presentation 11/03/2005Pavan KumarОценок пока нет
- Introduction To Bioinformatics: CSCI8980: Applied Machine Learning in Computational BiologyДокумент24 страницыIntroduction To Bioinformatics: CSCI8980: Applied Machine Learning in Computational BiologyAsmaОценок пока нет
- The Next Generation of Genomic ResearchДокумент75 страницThe Next Generation of Genomic ResearchbookloverinОценок пока нет
- High Throughput Methods in Proteomics: David Wishart University of Alberta Edmonton, ABДокумент72 страницыHigh Throughput Methods in Proteomics: David Wishart University of Alberta Edmonton, ABChandrashekar Yethagadahalli LingegowdaОценок пока нет
- Genome AnnotationДокумент58 страницGenome Annotationronnny11Оценок пока нет
- Lva1 App6891 PDFДокумент33 страницыLva1 App6891 PDFabhishekОценок пока нет
- Plasmodium Vivax: Parasitemia Determination by Real-Time Quantitative PCR in Aotus MonkeysДокумент4 страницыPlasmodium Vivax: Parasitemia Determination by Real-Time Quantitative PCR in Aotus MonkeysAndres JarrinОценок пока нет
- 1 What Is BioinformaticsДокумент34 страницы1 What Is BioinformaticsLaxmikant KambleОценок пока нет
- Gene RegulationДокумент37 страницGene RegulationJonas T. HingcoОценок пока нет
- Proteomics 1Документ73 страницыProteomics 1ShreevarОценок пока нет
- Yeast Microarrays For Genome Wide Parallel Genetic and Gene Expression AnalysisДокумент6 страницYeast Microarrays For Genome Wide Parallel Genetic and Gene Expression AnalysisDaniela CostaОценок пока нет
- Introduction To Molecular BiologyДокумент23 страницыIntroduction To Molecular Biologykris_bt20029241Оценок пока нет
- Basic Principles in Bioinformatics: Understanding MicroarraysДокумент81 страницаBasic Principles in Bioinformatics: Understanding MicroarraysBrandon ArceОценок пока нет
- Genomics 1Документ172 страницыGenomics 1AsandaОценок пока нет
- Lecture 1Документ38 страницLecture 1Đức Huy NguyễnОценок пока нет
- Gene Structure and Identification: Genes and Genomes Orfs and More Consensus Sequences Gene FindingДокумент16 страницGene Structure and Identification: Genes and Genomes Orfs and More Consensus Sequences Gene FindingimaemОценок пока нет
- Large-Scale Analysis of Gene ExpressionДокумент27 страницLarge-Scale Analysis of Gene Expressionk.felgenhauer137Оценок пока нет
- Mol Bio in Paternity Testing & ForensicsДокумент22 страницыMol Bio in Paternity Testing & ForensicsRainie PhamОценок пока нет
- 03 Gene ExpressionДокумент45 страниц03 Gene ExpressionJagan Karthik SugumarОценок пока нет
- RNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsДокумент6 страницRNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsnareshОценок пока нет
- DNA MicroarraysДокумент39 страницDNA Microarrayshira jamilОценок пока нет
- DNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensОт EverandDNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensОценок пока нет
- Module 6 - Product Analytics - IA - v2Документ9 страницModule 6 - Product Analytics - IA - v2Swapnil GudmalwarОценок пока нет
- CL415 MidsemДокумент2 страницыCL415 MidsemSwapnil GudmalwarОценок пока нет
- CL415 Midsem PDFДокумент2 страницыCL415 Midsem PDFSwapnil GudmalwarОценок пока нет
- PART-SG and PART-RKM Should Be Answered Separate AnswerbooksДокумент4 страницыPART-SG and PART-RKM Should Be Answered Separate AnswerbooksSwapnil GudmalwarОценок пока нет
- Design of Thin Walled Vessels Under Internal PressureДокумент2 страницыDesign of Thin Walled Vessels Under Internal PressureSwapnil GudmalwarОценок пока нет
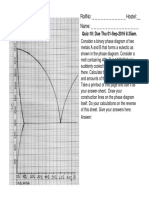
- Rollno: - Hostel: - NameДокумент1 страницаRollno: - Hostel: - NameSwapnil GudmalwarОценок пока нет
- PED AssignmentProblems 2017 PDFДокумент10 страницPED AssignmentProblems 2017 PDFSwapnil GudmalwarОценок пока нет
- CL409 Q3 PDFДокумент1 страницаCL409 Q3 PDFSwapnil GudmalwarОценок пока нет
- Quiz-8 (Take-Home Honour Code Applies) : Due 10.35 Am Mon 29-Aug-2016 Topic: X-Ray Crystallography (Self-Study Material Attached)Документ3 страницыQuiz-8 (Take-Home Honour Code Applies) : Due 10.35 Am Mon 29-Aug-2016 Topic: X-Ray Crystallography (Self-Study Material Attached)Swapnil GudmalwarОценок пока нет
- CL409 Handout 2 2016Документ13 страницCL409 Handout 2 2016Swapnil GudmalwarОценок пока нет
- CL409 Materials Science: Jayesh BellareДокумент12 страницCL409 Materials Science: Jayesh BellareSwapnil GudmalwarОценок пока нет
- Quiz-9: Due Tue 30-Aug-2016 11.35amДокумент1 страницаQuiz-9: Due Tue 30-Aug-2016 11.35amSwapnil GudmalwarОценок пока нет
- Quiz-5 (Take-Home Honour Code Applies) : Due 11.35 Am Tue 16-Aug-2016Документ2 страницыQuiz-5 (Take-Home Honour Code Applies) : Due 11.35 Am Tue 16-Aug-2016Swapnil GudmalwarОценок пока нет
- IIM LUCKNOW - Interview-Letter PDFДокумент2 страницыIIM LUCKNOW - Interview-Letter PDFSwapnil GudmalwarОценок пока нет
- BOCM 3714: T: +27 (0) 51 401 9111 - Info@ufs - Ac.za - WWW - Ufs.ac - ZaДокумент25 страницBOCM 3714: T: +27 (0) 51 401 9111 - Info@ufs - Ac.za - WWW - Ufs.ac - ZaNthabeleng NkaotaОценок пока нет
- Nuclei C Acids: BSN 1y0-5Документ25 страницNuclei C Acids: BSN 1y0-5joevette_30Оценок пока нет
- All Biology Questions and Answers F4 PDFДокумент35 страницAll Biology Questions and Answers F4 PDFmohammed jimjamОценок пока нет
- Tuesday 19 May 2020: Biology BДокумент28 страницTuesday 19 May 2020: Biology BAyaОценок пока нет
- Molecular Basis of Inheritance: By: Dr. Anand ManiДокумент130 страницMolecular Basis of Inheritance: By: Dr. Anand ManiIndu Yadav100% (3)
- Research Article: Involvement of Prohibitin Upregulation in Abrin-Triggered ApoptosisДокумент11 страницResearch Article: Involvement of Prohibitin Upregulation in Abrin-Triggered ApoptosisAndri Praja SatriaОценок пока нет
- Protein and The Element Sulfur: (No Need To Define)Документ3 страницыProtein and The Element Sulfur: (No Need To Define)Luis Gatchalian LacanilaoОценок пока нет
- Q3 Summex2 Sci10Документ1 страницаQ3 Summex2 Sci10Jeff Tristan CaliganОценок пока нет
- Letm19 BiosciДокумент16 страницLetm19 Bioscigretelabelong10Оценок пока нет
- Rajkumar Biology Printable Notes Unit 2 by Rajat.21-37Документ17 страницRajkumar Biology Printable Notes Unit 2 by Rajat.21-37Michael Benton100% (1)
- The Coronavirus Replicase: J. ZiebuhrДокумент38 страницThe Coronavirus Replicase: J. Ziebuhrfandangos presuntoОценок пока нет
- Principles of Genetics 6th Edition Snustad Test BankДокумент15 страницPrinciples of Genetics 6th Edition Snustad Test BankMelissaLeetioyq100% (18)
- Antibioticresistance PowerpointДокумент51 страницаAntibioticresistance PowerpointOdy100% (1)
- GTP BookДокумент113 страницGTP BookEDUARDOLOBOX100% (1)
- Protein Synthesis WorksheetДокумент4 страницыProtein Synthesis WorksheetJeff Nelson100% (2)
- DNA and Protein PowerpointДокумент13 страницDNA and Protein PowerpointKashish ShahОценок пока нет
- Cell Biology GlossaryДокумент55 страницCell Biology GlossaryEmrul HasanОценок пока нет
- J. Biol. Chem.-1989-Boggaram-11421-7Документ7 страницJ. Biol. Chem.-1989-Boggaram-11421-7Patrick RamosОценок пока нет
- First Aid MnemonicsДокумент27 страницFirst Aid MnemonicsRafael G. Garcia SanchezОценок пока нет
- Biology NTSE Stage-1Документ5 страницBiology NTSE Stage-1Sonal Gupta100% (5)
- Virus Structure and ReplicationДокумент42 страницыVirus Structure and Replicationtummalapalli venkateswara rao100% (2)
- Translation Dna To Mrna To ProteinДокумент5 страницTranslation Dna To Mrna To ProteinAna MarianaОценок пока нет
- Module 2. Molecular Biology (Week 4-7)Документ15 страницModule 2. Molecular Biology (Week 4-7)Nanette MangloОценок пока нет
- Genetic CodeДокумент36 страницGenetic CodeYashОценок пока нет
- Smooth and Granular Endoplasmic Reticulum RibosomesДокумент26 страницSmooth and Granular Endoplasmic Reticulum RibosomesArfiОценок пока нет
- Protein Synthesis ACEДокумент14 страницProtein Synthesis ACEZhiTing96Оценок пока нет
- RIBOSOMESДокумент10 страницRIBOSOMESJorge BazánОценок пока нет
- Lecture 36 - Translation and Protein TargetingДокумент24 страницыLecture 36 - Translation and Protein TargetingFarisha SultanОценок пока нет
- TranslationДокумент78 страницTranslationdrmukhtiarbaigОценок пока нет
- TB Final2023 643ef57734b858.643ef57a5a0fb6.40844090Документ71 страницаTB Final2023 643ef57734b858.643ef57a5a0fb6.40844090Sameh NoorОценок пока нет