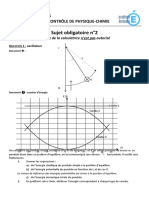
Вам также может понравиться
- Corrigé Série 1 TD UV-VIДокумент4 страницыCorrigé Série 1 TD UV-VIMuhammed Addala100% (3)
- Methodes de Calcul de RadiersДокумент35 страницMethodes de Calcul de Radierssahj95% (41)
- Chimie Organique Cours Sur Les AlcènesДокумент25 страницChimie Organique Cours Sur Les AlcènesKone KouweltonОценок пока нет
- CHAPITRE I Spectrophotométrie UV VisibleДокумент12 страницCHAPITRE I Spectrophotométrie UV VisibleMouka MoukaОценок пока нет
- Commissariat A L Énergie Atomique: Rapport CEAДокумент7 страницCommissariat A L Énergie Atomique: Rapport CEAfouad elferdiОценок пока нет
- Mat閞iaux Di閘ectriques - comportement Aux Champs 閘Документ36 страницMat閞iaux Di閘ectriques - comportement Aux Champs 閘joe kanikiОценок пока нет
- Electrochimie-Chapitre 1-R-KIHALДокумент15 страницElectrochimie-Chapitre 1-R-KIHALManar Smith100% (1)
- Devoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2014-2015) MR FKIRI FAOUZIДокумент4 страницыDevoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2014-2015) MR FKIRI FAOUZIMohamed SaidiОценок пока нет
- Spectre Atomique Cours 3 PDFДокумент4 страницыSpectre Atomique Cours 3 PDFMoufida ZouaghiОценок пока нет
- LemogifanirexibogejДокумент2 страницыLemogifanirexibogejVadel SniperОценок пока нет
- Bac Blanc N2 2009Документ12 страницBac Blanc N2 2009Amine NouarОценок пока нет
- Chimie C Chap10 Les PilesДокумент7 страницChimie C Chap10 Les PilesImed LatrechОценок пока нет
- Chimie Ds 1Документ2 страницыChimie Ds 1[AE]Оценок пока нет
- Devoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2011-2012) MR ALIBI ANOUARДокумент5 страницDevoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2011-2012) MR ALIBI ANOUARMohamed SaidiОценок пока нет
- (4 Points) : Niveau: Première S2 Composition Du Second Semestre Epreuve: Sciences Physiques 2018-2019 Durée: 03 HДокумент2 страницы(4 Points) : Niveau: Première S2 Composition Du Second Semestre Epreuve: Sciences Physiques 2018-2019 Durée: 03 HSYDIA ProdОценок пока нет
- 1cm44av9o 109949Документ3 страницы1cm44av9o 109949ndiayeОценок пока нет
- Pages de Bac Blanc 2Документ5 страницPages de Bac Blanc 2Khaouda Driss100% (1)
- Cours 2 Theorie Des Alliages Binaires Diagrammes D'équilibreДокумент21 страницаCours 2 Theorie Des Alliages Binaires Diagrammes D'équilibreslimane taleb bahmedОценок пока нет
- Contrôle Atomistique 1Документ4 страницыContrôle Atomistique 1[AE]100% (3)
- Serie-1-2022-23 (1)Документ8 страницSerie-1-2022-23 (1)mariem OuriОценок пока нет
- Spectre CoursДокумент7 страницSpectre CoursJerbi InesОценок пока нет
- Cap AtomistiqueДокумент40 страницCap AtomistiqueAll AhmeDciaОценок пока нет
- Expose - Techniques de Mesure de La Vitesse de CorrosionДокумент11 страницExpose - Techniques de Mesure de La Vitesse de CorrosionFlorian HonfinОценок пока нет
- Eoc 2Документ4 страницыEoc 2WilfriedОценок пока нет
- Spectroscopie ultra violet et VisibleДокумент10 страницSpectroscopie ultra violet et VisibleGhodbane AchrafОценок пока нет
- Cours 3 - Courant Électrique ContinuДокумент7 страницCours 3 - Courant Électrique ContinuMouna LaàtarОценок пока нет
- Devoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2014-2015) MR Handoura NaceurДокумент4 страницыDevoir de Synthèse N°3 - Sciences Physiques - Bac Sciences Exp (2014-2015) MR Handoura NaceurMohamed SaidiОценок пока нет
- Seance #6Документ19 страницSeance #6Mazama-Esso Moddoh OclooОценок пока нет
- Titrage AmpérométriqueДокумент13 страницTitrage AmpérométriqueSellam AnisОценок пока нет
- Cours PDFДокумент44 страницыCours PDFAdnen LaamouriОценок пока нет
- Spectrometrie de MasseДокумент8 страницSpectrometrie de MassesafemindОценок пока нет
- DSC2 2015 - 2016Документ14 страницDSC2 2015 - 2016boubacarbah054436Оценок пока нет
- Chapitre IIIДокумент10 страницChapitre IIIMED IMED-EDDINE SEGHIERОценок пока нет
- Geometrie Des MoleculesДокумент4 страницыGeometrie Des MoleculesqmwebtvqjlОценок пока нет
- Stabilité Des Éléments Chimiques ResuméДокумент2 страницыStabilité Des Éléments Chimiques Resuméعبد الفتاح الشقراويОценок пока нет
- Electrochimie 2010Документ74 страницыElectrochimie 2010kkОценок пока нет
- 02 - Spectrométrie UV - VisibleДокумент103 страницы02 - Spectrométrie UV - VisibleMalki kawtarОценок пока нет
- C1 - 02 - Les Atomes PolyélectroniquesДокумент2 страницыC1 - 02 - Les Atomes PolyélectroniquesKais Ben AichaОценок пока нет
- En PC Biof 2012 SRДокумент6 страницEn PC Biof 2012 SREL MEHDI EL HAMDOUCHIОценок пока нет
- PorrositéДокумент44 страницыPorrositéAbdo AiaicheОценок пока нет
- Chapitre II Uv VisibleДокумент10 страницChapitre II Uv VisibleA100% (1)
- Joussour-PC-7M-SN vf %281%29Документ172 страницыJoussour-PC-7M-SN vf %281%29pdpkwzz7mpОценок пока нет
- CoursДокумент44 страницыCoursAОценок пока нет
- Piles Electrochimiques PDFДокумент12 страницPiles Electrochimiques PDFAzizElheni100% (1)
- TDEEG0304Документ9 страницTDEEG0304Prince Abdoo100% (2)
- Chapitre IДокумент9 страницChapitre IŠãnFÖo Gh'aaОценок пока нет
- Chap3 Cours Electro MasterДокумент13 страницChap3 Cours Electro MasterLatifa MakhloufОценок пока нет
- Transformations Spontanees Dans Les Piles Et Production D Energie Resume de Cours 1 1Документ2 страницыTransformations Spontanees Dans Les Piles Et Production D Energie Resume de Cours 1 1Amine AlaoUii AlaouiОценок пока нет
- Mécanismes RéactionnelsДокумент3 страницыMécanismes RéactionnelsMorgan JaavuoОценок пока нет
- Interaction PDFДокумент42 страницыInteraction PDFmohamed benlarague100% (2)
- chp1 CEF 2023Документ78 страницchp1 CEF 2023mohaОценок пока нет
- Série Révision - Dipole RC-RLC-RLC Forcée - MR Mtibaa - PDF ( (Chap 1) ) - SfaxДокумент17 страницSérie Révision - Dipole RC-RLC-RLC Forcée - MR Mtibaa - PDF ( (Chap 1) ) - SfaxLifi SamОценок пока нет
- Cours Master FsbtairiДокумент95 страницCours Master Fsbtairilbenmok100% (3)
- Electronica Teoria de Circuitos y DispositivosДокумент58 страницElectronica Teoria de Circuitos y DispositivosJose Isaac Tuñoque GutiérrezОценок пока нет
- Conductimetrie TP 2 PDFДокумент16 страницConductimetrie TP 2 PDFMikel Carter83% (6)
- Chap1 Rappel Sur Les Solutions ÉlectrolytiquesДокумент9 страницChap1 Rappel Sur Les Solutions Électrolytiquesemma VОценок пока нет
- Cours de Chimie Organique Licence 1 Semestre 1Документ32 страницыCours de Chimie Organique Licence 1 Semestre 1Balla SangareОценок пока нет
- SMP 3 PDFДокумент60 страницSMP 3 PDFsoufyane el ouahabiОценок пока нет
- Devoir de Synthèse N°3 2ème Semestre - Sciences Physiques - Bac Sciences Exp (2018-2019) MR Hidri LazharДокумент4 страницыDevoir de Synthèse N°3 2ème Semestre - Sciences Physiques - Bac Sciences Exp (2018-2019) MR Hidri LazharMohamed SaidiОценок пока нет
- ts1_partie_1_chimie_le_symbolisme_des_fleches_courbesДокумент4 страницыts1_partie_1_chimie_le_symbolisme_des_fleches_courbesmaxence.boronat2006Оценок пока нет
- ChloreДокумент18 страницChloreDarel NadjieraОценок пока нет
- UraniumДокумент7 страницUraniumAbdelkader WalidОценок пока нет
- StereochimieДокумент17 страницStereochimieDarel NadjieraОценок пока нет
- Proprietés Physique Meca Des MateriauxДокумент206 страницProprietés Physique Meca Des Materiauxabderazak_2008100% (1)
- Spectro IRДокумент14 страницSpectro IRDarel NadjieraОценок пока нет
- CCMДокумент4 страницыCCMDarel NadjieraОценок пока нет
- ChromatoДокумент10 страницChromatorimbrahimОценок пока нет
- SoufreДокумент8 страницSoufreDarel NadjieraОценок пока нет
- MetauxДокумент13 страницMetauxDarel NadjieraОценок пока нет
- NomenclatureДокумент7 страницNomenclatureDarel NadjieraОценок пока нет
- COR706Документ132 страницыCOR706raly13100% (1)
- Chimie OrganiqueДокумент266 страницChimie OrganiqueTram TruongОценок пока нет
- Chimie OrganiqueДокумент266 страницChimie OrganiqueTram TruongОценок пока нет
- Chimie OrganiqueДокумент266 страницChimie OrganiqueTram TruongОценок пока нет
- SiliciumДокумент7 страницSiliciumDarel NadjieraОценок пока нет
- Ewe Cours1Документ1 страницаEwe Cours1Darel NadjieraОценок пока нет
- OxygeneДокумент24 страницыOxygeneDarel NadjieraОценок пока нет
- ComplexesДокумент6 страницComplexesDarel NadjieraОценок пока нет
- MetauxДокумент13 страницMetauxDarel NadjieraОценок пока нет
- HPLCДокумент10 страницHPLCadribispo100% (1)
- HuckelДокумент13 страницHuckelDarel NadjieraОценок пока нет
- Liaison CovalenteДокумент19 страницLiaison CovalenteDarel NadjieraОценок пока нет
- Derives HalogenДокумент29 страницDerives Halogenhijim371Оценок пока нет
- FerДокумент14 страницFerDarel NadjieraОценок пока нет
- CPGДокумент6 страницCPGDarel NadjieraОценок пока нет
- ChromatoДокумент10 страницChromatorimbrahimОценок пока нет
- GlucidesДокумент11 страницGlucidesDarel NadjieraОценок пока нет
- ChloreДокумент18 страницChloreDarel NadjieraОценок пока нет
- Derives HalogenДокумент29 страницDerives Halogenhijim371Оценок пока нет
- CCMДокумент4 страницыCCMDarel NadjieraОценок пока нет
- Decret 15-76Документ4 страницыDecret 15-76Cris GauchoОценок пока нет
- TP Manip Elem Bio Mol TechДокумент3 страницыTP Manip Elem Bio Mol TechDr.Zakaria MFTОценок пока нет
- Chapitre II Les Équipements Pour L'intensification Des ProcédésДокумент10 страницChapitre II Les Équipements Pour L'intensification Des ProcédésRay CharlesОценок пока нет
- EXP-MN-SI170-FR-R0 - Principales Pannes PDFДокумент62 страницыEXP-MN-SI170-FR-R0 - Principales Pannes PDFbali100% (6)
- Le NoyauДокумент104 страницыLe NoyauOussamaMafiousiОценок пока нет
- Chapitre I Caracteristiques Mecaniques Des Materiaux & Hypotheses de CalculДокумент8 страницChapitre I Caracteristiques Mecaniques Des Materiaux & Hypotheses de Calculadmine100% (2)
- Forces PressantesДокумент24 страницыForces PressantesLagdaa MohammedОценок пока нет
- Cours La Cinétique FormelleДокумент7 страницCours La Cinétique Formellesafae ziyatiОценок пока нет
- Durete Totale D'une EauДокумент8 страницDurete Totale D'une EauazebelazebelОценок пока нет
- 2 These JuillardMДокумент231 страница2 These JuillardMMohammed KouidriОценок пока нет
- ARCHE - Ancrage Eurocode 2 PDFДокумент6 страницARCHE - Ancrage Eurocode 2 PDFrodrigowinkОценок пока нет
- Pds Hempadur Zinc 17340 FR-FRДокумент3 страницыPds Hempadur Zinc 17340 FR-FRKhaled bouhlelОценок пока нет
- Devoir 1 Modele 1 Physique Chimie 2 Bac SM Semestre 1Документ4 страницыDevoir 1 Modele 1 Physique Chimie 2 Bac SM Semestre 1Walid ErraОценок пока нет
- Processus de Dessalage Du Petrole À La SorazДокумент46 страницProcessus de Dessalage Du Petrole À La SorazMamane Bachir80% (5)
- Destruction Du Cyanure SGSДокумент3 страницыDestruction Du Cyanure SGSrolandoh1Оценок пока нет
- Memoire de Magister MEZARI NAOUELДокумент125 страницMemoire de Magister MEZARI NAOUELgfgf100% (2)
- ArchaeaДокумент205 страницArchaeaBoudyachОценок пока нет
- Devoir Synthése 1 2em 2020Документ2 страницыDevoir Synthése 1 2em 2020Chaimma KalaouiОценок пока нет
- Cours de Chimie Verte, AnДокумент7 страницCours de Chimie Verte, AnTaim KhouriОценок пока нет
- Propmeca Etu2Документ8 страницPropmeca Etu2Rimas InconueОценок пока нет
- CC-Exos 2009-2010 2 PDFДокумент10 страницCC-Exos 2009-2010 2 PDFHamza BoulikaОценок пока нет
- PNM 10.1.706Документ27 страницPNM 10.1.706Younes El-BouznaniОценок пока нет
- RimalДокумент1 страницаRimalOmar OuchaneОценок пока нет
- ETD0236c - Principes Supportage de TuyauteriesДокумент6 страницETD0236c - Principes Supportage de TuyauteriesingetecmcОценок пока нет
- Robinet A SoufflerДокумент1 страницаRobinet A SoufflerMars76Оценок пока нет
- EPREUVE DE PCT 6ieme Séquence 3eДокумент2 страницыEPREUVE DE PCT 6ieme Séquence 3ePrince NgaleОценок пока нет
- Puscarzyk E. Aspects Toxicocinétiques, Toxicodynamiques Et ThérapeutiquesДокумент220 страницPuscarzyk E. Aspects Toxicocinétiques, Toxicodynamiques Et ThérapeutiquesniebelungenОценок пока нет
- Genie GenetiqueДокумент107 страницGenie GenetiqueYousfi Koussaila80% (10)
- Procdure Travail Espace Clos-UQAMДокумент19 страницProcdure Travail Espace Clos-UQAMElmahjoub LAGRINIОценок пока нет