Вам также может понравиться
- Dmt.4 ChlorobutanalДокумент4 страницыDmt.4 ChlorobutanalMonique Reina LagartijaОценок пока нет
- Lai2002 PDFДокумент10 страницLai2002 PDFHimanshuОценок пока нет
- Conversion of Seismic WavesДокумент3 страницыConversion of Seismic WavesMaliha N. AhmedОценок пока нет
- Ylva E. Bergman, Roger Mulder and Patrick Perlmutter - Total Synthesis of 20-Norsalvinorin A. 1. Preparation of A Key IntermediateДокумент4 страницыYlva E. Bergman, Roger Mulder and Patrick Perlmutter - Total Synthesis of 20-Norsalvinorin A. 1. Preparation of A Key IntermediateBic0000Оценок пока нет
- 5 Epi LiphagalДокумент3 страницы5 Epi Liphagalglreddy09Оценок пока нет
- Tetracycline HarvardДокумент2 страницыTetracycline HarvardanisarizcaОценок пока нет
- Smith Transannular DAДокумент4 страницыSmith Transannular DASatyaki MajumdarОценок пока нет
- Trans Imminazione Beta ControllataДокумент2 страницыTrans Imminazione Beta ControllatafuturichimiciОценок пока нет
- Regioselective Formylation of Pyrazolo (3,4-b) Pyridine and Pyrazolo (1,5-A) Pyrimidine Systems Using Vilsmeier-Haack ConditionsДокумент3 страницыRegioselective Formylation of Pyrazolo (3,4-b) Pyridine and Pyrazolo (1,5-A) Pyrimidine Systems Using Vilsmeier-Haack ConditionsJorge L MuñozОценок пока нет
- Enantioselective in AllylationДокумент4 страницыEnantioselective in Allylationredevol7Оценок пока нет
- Biginille Reaction Mechanism PDFДокумент4 страницыBiginille Reaction Mechanism PDFshenn0Оценок пока нет
- J. Org. Chem. 2003, 68 (17), 6788-6970Документ3 страницыJ. Org. Chem. 2003, 68 (17), 6788-6970srinivasarao meneniОценок пока нет
- Jo 0503299Документ6 страницJo 0503299Kyucheol PaikОценок пока нет
- Synth-Of Azitrhomycin New ProcedureДокумент3 страницыSynth-Of Azitrhomycin New ProcedureMario MicciarelliОценок пока нет
- Joc - 7099Документ8 страницJoc - 7099Diogo DiasОценок пока нет
- Two-Step Synthesis of Aza-And Diazaindoles From Chloroamino - N-Heterocycles Using EthoxyvinylborolaneДокумент5 страницTwo-Step Synthesis of Aza-And Diazaindoles From Chloroamino - N-Heterocycles Using EthoxyvinylborolaneSilvanaMedhatОценок пока нет
- HecogeninaДокумент4 страницыHecogeninaFernando Torres PérezОценок пока нет
- Joc - 7085Документ7 страницJoc - 7085Diogo DiasОценок пока нет
- 303 PDFДокумент12 страниц303 PDFSilvanaMedhatОценок пока нет
- Polyphosphoric Acid Trimethylsilyl Ester Promoted Intramolecular Acylation of An Olefin by A Carboxylic AcidДокумент4 страницыPolyphosphoric Acid Trimethylsilyl Ester Promoted Intramolecular Acylation of An Olefin by A Carboxylic AcidrosanaОценок пока нет
- Thalidomide From Liquid AmmoniaДокумент4 страницыThalidomide From Liquid AmmoniaFurphy1Оценок пока нет
- Total Synthesis of ( - Deoxypenostatin A. Approaches To The Syntheses of Penostatins A and BДокумент9 страницTotal Synthesis of ( - Deoxypenostatin A. Approaches To The Syntheses of Penostatins A and BrrgodboleОценок пока нет
- Heteroaryl Cross-Coupling As An Entry Toward The Synthesis of Lavendamycin Analogues: A Model StudyДокумент10 страницHeteroaryl Cross-Coupling As An Entry Toward The Synthesis of Lavendamycin Analogues: A Model StudySilvanaMedhatОценок пока нет
- ( ) AspidospermidineДокумент3 страницы( ) AspidospermidineLing LingОценок пока нет
- A Short Total Synthesis of - Aspidospermidine: Lisa A. Sharp and Samir Z. ZardДокумент4 страницыA Short Total Synthesis of - Aspidospermidine: Lisa A. Sharp and Samir Z. ZardMarius ConstantinОценок пока нет
- Total Synthesis of Sordaricin: Lewis N. Mander and Regan J. ThomsonДокумент4 страницыTotal Synthesis of Sordaricin: Lewis N. Mander and Regan J. ThomsonOskar Martin OrdoñezОценок пока нет
- Facile Synthesis of O - Alkyl-, O - Aryl-, and Diaminopurine Nucleosides From 2 - DeoxyguanosineДокумент4 страницыFacile Synthesis of O - Alkyl-, O - Aryl-, and Diaminopurine Nucleosides From 2 - DeoxyguanosineWalid EbaiedОценок пока нет
- 3 Bcnzylidenc 2,5 DiketopiperazincДокумент2 страницы3 Bcnzylidenc 2,5 DiketopiperazincgeliliОценок пока нет
- Molbank: Synthesis and Characterization of A Novel 2-PyrazolineДокумент4 страницыMolbank: Synthesis and Characterization of A Novel 2-PyrazolineAndre BertuahОценок пока нет
- Cyclization of R-Oxo-Oximes To 2-Substituted BenzoxazolesДокумент7 страницCyclization of R-Oxo-Oximes To 2-Substituted Benzoxazolespaula salamancaОценок пока нет
- Oxidation of Indoles To Isatins OL 2014Документ4 страницыOxidation of Indoles To Isatins OL 2014Victor CiocalteaОценок пока нет
- Twofold CH Functionalization: Palladium-Catalyzed Ortho Arylation of AnilidesДокумент5 страницTwofold CH Functionalization: Palladium-Catalyzed Ortho Arylation of Anilidesmalala000Оценок пока нет
- Ol 052578 PДокумент4 страницыOl 052578 PChitrangadha ReddyОценок пока нет
- Can. J. Chem. 49, 1071-1084 (1971) - Methyl 4-BromobutyrateДокумент15 страницCan. J. Chem. 49, 1071-1084 (1971) - Methyl 4-Bromobutyratesunil_vaman_joshiОценок пока нет
- Carrira GlycosideДокумент3 страницыCarrira GlycosideYasser SamirОценок пока нет
- 2005 Organometallics 2005 Artículo DelДокумент9 страниц2005 Organometallics 2005 Artículo DelItzel MercadoОценок пока нет
- Catalytic C H Functionalization Driven by CO As A Stoichiometric Reductant: Application To Carbazole SynthesisДокумент3 страницыCatalytic C H Functionalization Driven by CO As A Stoichiometric Reductant: Application To Carbazole SynthesisJORGE IVAN CASTRO CASTROОценок пока нет
- B 3Документ2 страницыB 3bijuarОценок пока нет
- Improved Synthesis of Enantiopure 4-Hydroxy (2.2) ParacyclophaneДокумент3 страницыImproved Synthesis of Enantiopure 4-Hydroxy (2.2) ParacyclophaneDiogo DiasОценок пока нет
- Enantioselective Synthesis of Dihydropyridinones Via NHC-Catalyzed Aza-Claisen ReactionДокумент4 страницыEnantioselective Synthesis of Dihydropyridinones Via NHC-Catalyzed Aza-Claisen ReactionlucerocaОценок пока нет
- Synthesis of Schiff Bases Using Greener MethodologiesДокумент5 страницSynthesis of Schiff Bases Using Greener MethodologiesDownload_from_ScribdОценок пока нет
- 2005 OrgLett Highly Chemoselective Addition of Amines To Epoxides in WaterДокумент3 страницы2005 OrgLett Highly Chemoselective Addition of Amines To Epoxides in Waterjames mellaleievОценок пока нет
- THE STRUCTURE OF METOPON - J. Am. Chem. Soc., 1953, 75 (17), PP 4373-4374Документ2 страницыTHE STRUCTURE OF METOPON - J. Am. Chem. Soc., 1953, 75 (17), PP 4373-4374muopioidreceptorОценок пока нет
- Cocaine Synthesis Shing Org Lett 2011Документ4 страницыCocaine Synthesis Shing Org Lett 2011Cody DunnОценок пока нет
- Masato Koreeda, Lindsey Brown and Leander J. Valdes III - The Absolute Stereochemistry of SalvinorinsДокумент4 страницыMasato Koreeda, Lindsey Brown and Leander J. Valdes III - The Absolute Stereochemistry of SalvinorinsnnnnjwОценок пока нет
- Comparison The Reactivity S - Adenylic Acid and S - Guanylic AcidДокумент5 страницComparison The Reactivity S - Adenylic Acid and S - Guanylic AcidEr Mayur PatilОценок пока нет
- Organic Letters (2008), 10 (17), 3907-3909Документ3 страницыOrganic Letters (2008), 10 (17), 3907-3909James TianОценок пока нет
- Riboceine Paper 20 1Документ6 страницRiboceine Paper 20 1api-257130539Оценок пока нет
- 5 - Methylaristeromycin and Related Derivatives: Wei Ye and Stewart W. SchnellerДокумент3 страницы5 - Methylaristeromycin and Related Derivatives: Wei Ye and Stewart W. Schnellermanil_mohanОценок пока нет
- Green Chemistry Approach To The Synthesis of N-Substituted Piperidones PDFДокумент3 страницыGreen Chemistry Approach To The Synthesis of N-Substituted Piperidones PDFjavasoloОценок пока нет
- An Efficient Synthesis of 5,7-Dimethoxy-4-Methylphthalide, A Key Intermediate in The Synthesis of Mycophenolic AcidДокумент2 страницыAn Efficient Synthesis of 5,7-Dimethoxy-4-Methylphthalide, A Key Intermediate in The Synthesis of Mycophenolic AcidAmeerul HazeeqОценок пока нет
- Synthesis of Analogues of 1,3-Dihydroxyacetone Phosphate and Glyceraldehyde 3-Phosphate For Use in Studies of Fructose-1,6-Diphosphate Aldolase'Документ9 страницSynthesis of Analogues of 1,3-Dihydroxyacetone Phosphate and Glyceraldehyde 3-Phosphate For Use in Studies of Fructose-1,6-Diphosphate Aldolase'Carlos MRgzОценок пока нет
- J. Org. Chem. 19A New Method For The Synthesis of Heptamethine Cyanine Dyes: Synthesis of New Near-Infrared Fluorescent Labels5,60, 2391-2395 2391Документ5 страницJ. Org. Chem. 19A New Method For The Synthesis of Heptamethine Cyanine Dyes: Synthesis of New Near-Infrared Fluorescent Labels5,60, 2391-2395 2391kawtherahmedОценок пока нет
- JOC 2009 74 (19) p7556-7558Документ3 страницыJOC 2009 74 (19) p7556-7558고 영옥Оценок пока нет
- Sdarticle 4Документ3 страницыSdarticle 4venkigitamОценок пока нет
- Ullmann Acridine SynthesisДокумент4 страницыUllmann Acridine SynthesisChiến NguyễnОценок пока нет
- 1057Документ5 страниц1057Amer KasidehОценок пока нет
- Efficient Synthesis of 1,5-BenzodiazepinesДокумент3 страницыEfficient Synthesis of 1,5-BenzodiazepinesdoubleffectОценок пока нет
- Syntheses of The Enantiomers of 1-Deoxynojirimycin and 1-Deoxyaltronojirimycin Via Chemo-And Diastereoselective Olefinic Oxidation of Unsaturated AminesДокумент14 страницSyntheses of The Enantiomers of 1-Deoxynojirimycin and 1-Deoxyaltronojirimycin Via Chemo-And Diastereoselective Olefinic Oxidation of Unsaturated AminesDiogo DiasОценок пока нет
- 2009 JOC AziridinesДокумент8 страниц2009 JOC AziridinesCarmen SimalОценок пока нет
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesОт EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesОценок пока нет
- Cryogenic Pressure Temperature Swing Adsorption Process For Natural Gas UpgradeДокумент2 страницыCryogenic Pressure Temperature Swing Adsorption Process For Natural Gas UpgradekevinОценок пока нет
- BOQ For Dubti Dam-GeotecДокумент4 страницыBOQ For Dubti Dam-GeotecMehari GebremeskelОценок пока нет
- Marshall's Reagent: Origins, Modifications, and New ApplicationsДокумент2 страницыMarshall's Reagent: Origins, Modifications, and New Applicationso_l_0Оценок пока нет
- Superhydrophobic Carbon-Based Materials: A Review of Synthesis, Structure, and ApplicationsДокумент16 страницSuperhydrophobic Carbon-Based Materials: A Review of Synthesis, Structure, and ApplicationsAnirban RoyОценок пока нет
- Navarro, Juan Miguel v. - Plan413 - Asynchronous Activity 2Документ4 страницыNavarro, Juan Miguel v. - Plan413 - Asynchronous Activity 2Juan Miguel NavarroОценок пока нет
- Liquid Nitrogen As A Non Polluting FuelДокумент30 страницLiquid Nitrogen As A Non Polluting FuelShubham Raghuvanshi100% (2)
- Star Chart June 2022Документ1 страницаStar Chart June 2022Honolulu Star-AdvertiserОценок пока нет
- Conectores PostesДокумент2 страницыConectores PostesHabiran GonzalezОценок пока нет
- General Monitors S5000 Gas Detector configuration-ENДокумент2 страницыGeneral Monitors S5000 Gas Detector configuration-ENShahebaz KhanОценок пока нет
- Quiz No. 1Документ1 страницаQuiz No. 1Juanito Jr OriginesОценок пока нет
- Free-Fall Practice ProblemsДокумент1 страницаFree-Fall Practice ProblemsGabriel Muñiz NegrónОценок пока нет
- Laccase Immobilization For Water Purification - A Comprehensive ReviewДокумент15 страницLaccase Immobilization For Water Purification - A Comprehensive ReviewgustavoОценок пока нет
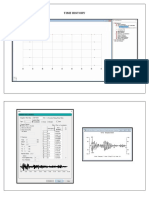
- (UPDATED) Time HistoryДокумент10 страниц(UPDATED) Time HistoryShaina Mariz PanaliganОценок пока нет
- EN J02.DAI.02 Daikin Sensira RXF C B A Technical Data RXF C Data BookДокумент26 страницEN J02.DAI.02 Daikin Sensira RXF C B A Technical Data RXF C Data BookolafОценок пока нет
- 4july BookДокумент5 страниц4july BookDevansh AggarwalОценок пока нет
- Mma 070921 Endress Liquidlevelpart1Документ4 страницыMma 070921 Endress Liquidlevelpart1sarsureshОценок пока нет
- 5.17 - A10706002 - Lely Ayu NingsihДокумент8 страниц5.17 - A10706002 - Lely Ayu NingsihLely Ayu NingsihОценок пока нет
- Chapter 2.6 Aldehyde & KetoneДокумент40 страницChapter 2.6 Aldehyde & Ketone0JTINGОценок пока нет
- Physics 202 Experiment #7 Diffraction Grating Pre-LabДокумент6 страницPhysics 202 Experiment #7 Diffraction Grating Pre-LabDennis LingОценок пока нет
- Grade 6 DLL SCIENCE 6 Q3 Week 6Документ5 страницGrade 6 DLL SCIENCE 6 Q3 Week 6Mark neil a. GalutОценок пока нет
- Atomic and Molecular Physics (PDFDrive)Документ103 страницыAtomic and Molecular Physics (PDFDrive)Ajay MinhasОценок пока нет
- SBM Schiedam Compressor Piping DesignДокумент45 страницSBM Schiedam Compressor Piping DesignSeudonim SatoshiОценок пока нет
- Sugar IndustriesДокумент46 страницSugar Industriesshine king100% (1)
- 12.1.5 Atomic Structure Electron ConfigurationДокумент54 страницы12.1.5 Atomic Structure Electron ConfigurationboobooОценок пока нет
- C5RA01911GДокумент55 страницC5RA01911GAndrew LondonОценок пока нет
- Material Safety Data SheetДокумент7 страницMaterial Safety Data SheetAnonymous NgcpLQiОценок пока нет
- Labsheet 3 - F2FДокумент9 страницLabsheet 3 - F2FMuhammad ZahidОценок пока нет
- MPPL PT Calendar 2024 2025Документ6 страницMPPL PT Calendar 2024 2025Joby GeorgeОценок пока нет
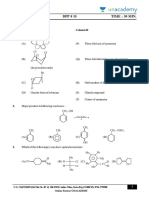
- DPP # 1 5 Time: 30 Min.: - Column I Column IIДокумент3 страницыDPP # 1 5 Time: 30 Min.: - Column I Column IIAkhil JamwalОценок пока нет