Вам также может понравиться
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5795)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (345)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Formulation and Calculation of Isoparametric Finite Element MatrixДокумент27 страницFormulation and Calculation of Isoparametric Finite Element MatrixSanjib RaiОценок пока нет
- Steam Turbine: Bearings Wheels and DiaphragmsДокумент128 страницSteam Turbine: Bearings Wheels and DiaphragmsJeeEianYann100% (1)
- NaphthaleneДокумент3 страницыNaphthaleneNur Hafeza75% (4)
- Helically Coiled TubesДокумент11 страницHelically Coiled TubesBenОценок пока нет
- FEF Welding Fabric Selection ChartДокумент1 страницаFEF Welding Fabric Selection Chartsalman KhanОценок пока нет
- Mech Eng DsДокумент11 страницMech Eng Ds路人丁Оценок пока нет
- Instruction: Encircle The Correct Answer. Erasures Are Considered Wrong. Test I: Multiple ChoiceДокумент4 страницыInstruction: Encircle The Correct Answer. Erasures Are Considered Wrong. Test I: Multiple ChoiceVenece Abellana0% (1)
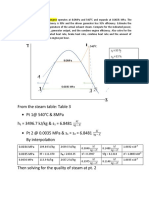
- From The Steam Table: Table 3 PT 1@ 540 C & 8mpa H 3496.7 KJ/KG & S 6.8481 PT 2 at 0.0035 Mpa & S S 6.8481 by InterpolationДокумент5 страницFrom The Steam Table: Table 3 PT 1@ 540 C & 8mpa H 3496.7 KJ/KG & S 6.8481 PT 2 at 0.0035 Mpa & S S 6.8481 by InterpolationMark AgusОценок пока нет
- Exp 3 Plate Heat ExchangerДокумент6 страницExp 3 Plate Heat ExchangerMeema FaatimahbagОценок пока нет
- Desalination: P. Palomar, J.L. Lara, I.J. LosadaДокумент15 страницDesalination: P. Palomar, J.L. Lara, I.J. Losadajean miguel oscorima celisОценок пока нет
- Letter To Editor On The Effects of Climate Change On The EnvironmentДокумент2 страницыLetter To Editor On The Effects of Climate Change On The EnvironmentKAYANNA LAWRENCEОценок пока нет
- Fluid Mechanics II (Chapter4)Документ15 страницFluid Mechanics II (Chapter4)حيدر محمدОценок пока нет
- Charny - Mathematical Models of Bioheat TransferДокумент137 страницCharny - Mathematical Models of Bioheat TransferMadalena PanОценок пока нет
- Pressure: Unit 2Документ14 страницPressure: Unit 2Sumit ShahОценок пока нет
- Effect of Impurities On TheДокумент6 страницEffect of Impurities On TheBansal Shivansh100% (1)
- Heat and Mass TransferДокумент31 страницаHeat and Mass TransferRavi RaneОценок пока нет
- Creep Behavior of Metals/AlloysДокумент2 страницыCreep Behavior of Metals/AlloysNitinSrivastavaОценок пока нет
- Pre Board - Science - Class 10 QPДокумент5 страницPre Board - Science - Class 10 QP10E Yuvan Sarabeshan Thirumeninathan [3383]Оценок пока нет
- Clo 7Документ42 страницыClo 7Shahadat AwanОценок пока нет
- Lec13 Problem SolutionДокумент30 страницLec13 Problem Solutionbadviolenceisbad100% (1)
- Explosive WeldingДокумент3 страницыExplosive WeldingDarryl007Оценок пока нет
- Coh FRANC2D Tutorial PCДокумент0 страницCoh FRANC2D Tutorial PCthegreatest0888Оценок пока нет
- Theory of Irregular Impedance Waveguides, Generalised Method of Separation of Variables - Zaginaylov Et Al (MMET 2014)Документ6 страницTheory of Irregular Impedance Waveguides, Generalised Method of Separation of Variables - Zaginaylov Et Al (MMET 2014)dpshepherdОценок пока нет
- Level Ii Questions - RTДокумент7 страницLevel Ii Questions - RTViral ThankiОценок пока нет
- RV College of EngineeringДокумент12 страницRV College of EngineeringRahul ChandraОценок пока нет
- 1204 0162Документ2 страницы1204 0162roar123Оценок пока нет
- Experiment 1: Heat Transfer Measurements Using A Thermal Imaging Infrared CameraДокумент6 страницExperiment 1: Heat Transfer Measurements Using A Thermal Imaging Infrared CamerapmnitsОценок пока нет
- Unit One Material and Geometry of Cutting Tools 2015Документ46 страницUnit One Material and Geometry of Cutting Tools 2015elnat feyisa100% (1)
- Damping Dissipation AbsorbtionДокумент3 страницыDamping Dissipation AbsorbtionLia MatiasОценок пока нет