Академический Документы
Профессиональный Документы
Культура Документы
CH 290
Загружено:
Silas LookenИсходное описание:
Оригинальное название
Авторское право
Доступные форматы
Поделиться этим документом
Поделиться или встроить документ
Этот документ был вам полезен?
Это неприемлемый материал?
Пожаловаться на этот документАвторское право:
Доступные форматы
CH 290
Загружено:
Silas LookenАвторское право:
Доступные форматы
04/11/2012 03:26
Molecular reaction dynamics
Collision Theory
6/2/2008
04/11/2012 03:26
Course Outline (2 lectures/w and 2Tutorial/w)
Microscopic theories of reaction rates: Collision and transition state
theories
Elementary reactions
Rates and mechanisms of chemical reactions (reactions rates and
equilibria, enzyme catalysed reactions)
Electromotive force and electrochemical cells
Electrolytes in solutions
The Debye-Huckel theory of interionic interaction and ionic activity
coefficient
CH 344: CHEMICAL KINETICS AND
ELECTROCHEMISTRY (2 units)
Dr. E.B. Mubofu and G. Kinunda
04/11/2012 03:26
ASSESSMENT MODE:
1. Set of questions to be distributed for the tutorials
2. There will be 2 tests, each carry 20 Marks
3. Final examination: 60 Marks
Section A: 30 Marks
Section B: 30 Marks, 3 Questions answer 2
References:
1. P.W. Atkins Physical chemistry (5th, 6th or 7th
ed.)
2. G.M. Barrow Physical chemistry 3rd ed. 1973 or
later ed.
3. B. G. Segal Chemistry experiment and theory, 1985
4. Any advanced physical chemistry book
04/11/2012 03:26
Molecular reaction dynamics
Collision Theory
6/2/2008
04/11/2012 03:26
The pharmacokinetics of artensunate is that
following oral administtration .. reaches Cmax
within 45 to 90 minutes. The product is
effective against malaria by the same mechanism
of action.
04/11/2012 03:26
You may recall that thermodynamics can predict whether
or not a reaction will proceed but cannot tell us how fast
or slow will the reaction occur! NOW
Chemical kinetics deals with the understanding of
the factors that affect the rate of a chemical
reaction and elucidates information about the
mechanism of the reaction
(A reaction mechanism is a series of steps that defines the order
in which bonds are broken and new ones are formed, until the
final product is obtained)
chemical reaction kinetics is concerned with the
rates at which chemical reactions occur
NB: Generally, a rate is a change in some quantity wrt time
04/11/2012 03:26
Nature of reactants: Each reaction proceeds at its own rate.
Changing the nature of reactants will, in general, change the
rate of the reaction
The concentration of reactants: For a chemical reaction to
occur, the reactants must come close together and collide.
Because collisions are more frequent when concentrations
increase, then the reaction rates increase as the concentration
of the reactants increase
The temperature: In general, increasing the temperature
increases the rate of the reaction. The rate of most reactions
increases by a factor of 2 or 3 for a 10
o
C rise in temperature!
The nature of the solvent:Although not all reactions occur in
solution, but for those carried in solution, changing the solvent
will generally change the rate of the reaction
The presence of the catalyst
What factors affect the rate of a chemical reaction?
04/11/2012 03:26
Molecular reaction dynamics
Why do reactions occur?
How?! i.e pathways and rate/speed
This is the heart and the cornerstone of chemistry
(why?) because here we examine the details of
what happens to molecules at the climax of
reactions and hence we can establish
mechanisms of reactions
The simplest quantitative account of reaction rates
at microscopic level is in terms of collision theory
which can be used only for discussion of reactions
between simple species in the gaseous phase
04/11/2012 03:26
04/11/2012 03:26
Collision Theory of bimolecular gas reaction
Generally, a theory is an explanation for
something which happens AND
Collision Theory is a theory that assumes
that, in order for a reaction to occur,
reactant molecules must COLLIDE with
an ENERGY GREATER than some
minimal value and with a PROPER
ORIENTATION
04/11/2012 03:26
Collision Theory
In other words, collision theory maintains that
the rate constant for a reaction is the product
of three factors.
1. Z, the collision frequency
2. f, the fraction of collisions with sufficient energy to
react
3. p, the fraction of collisions with the proper
orientation to react
4. Mathematically,
04/11/2012 03:26
Collision frequency
Z is only slightly temperature dependent.
This is illustrated using the kinetic theory of gases, which
shows the relationship between the velocity of gas
molecules and their absolute temperature.
or
04/11/2012 03:26
c o \T/M
This implies that the higher the temperature, the higher the root mean square
speed of the molecules, and at a given temperature heavy molecules travel
more slowly than light molecules
04/11/2012 03:26
- A collision will only be successful
only if the kinetic energy exceeds a
minimum energy value,
a
c
of the
reaction THUS
- The rate constant should also be
proportional to a Boltzmann factor
of the form
RT
E
a
e
, so we can
anticipate that
k
2
o o(\T/M)e
-Ea/RT
04/11/2012 03:26
-
Moreover, not every collision will
lead to reaction even if the energy
requirement is satisfied, because the
reactants may need to collide in a
certain orientation. The steric
requirement suggests that another
factor, p, should be introduced, and
thus k
2
o op(\T/M)e
-Ea/RT
- Indeed this is what
is predicted by the collision theory!
It reflects that for a successful reaction/effective
collision:-
k
2
o encounter rate x minimum energy x steric requirement
04/11/2012 03:26
Collision Theory-Collision energy
That is, f, the fraction of molecules with
sufficient threshold energy turns out to be
very temperature dependent.
This is the primary factor relating temperature increases to
observed rate increases.
04/11/2012 03:26
Collision Theory and reaction rates
The reaction rate dependence on the
fraction of collisions with the proper
orientation, p, is independent of
temperature changes
So, with changes in temperature, Z and p
remain fairly constant.
We can thus use that fact to derive a
mathematical relationship between the rate
constant, k, and the absolute
temperature.
04/11/2012 03:26
The Arrhenius Equation
If we were to combine the relatively
constant terms, Z and p, into one constant,
and call it A. We obtain the Arrhenius
equation:
The Arrhenius equation expresses the dependence of the
rate constant on absolute temperature and activation
energy.
04/11/2012 03:26
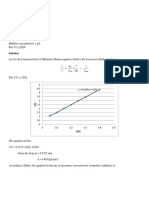
The Arrhenius Equation
It is useful to recast the Arrhenius equation
in logarithmic form.
We can relate this equation to the (somewhat rearranged) general
formula for a straight line.
y = b + m x
A plot of ln k versus (1/T) should yield a straight line with a slope
of (-E
a
/R) and an intercept of ln A.
04/11/2012 03:26
The Arrhenius Equation
A more useful form of the equation emerges
if we look at two points on the line this
equation describes that is, (k
1
, (1/T
1
)) and
(k
2
, (1/T
2
)).
With this form of the equation, given the activation
energy and the rate constant k
1
at a given
temperature T
1
, we can find the rate constant k
2
at any other temperature, T
2
.
04/11/2012 03:26
Now, let us compare k
2
= N
A
op(\T/M)e
-Ea/RT
with
04/11/2012 03:26
Note: The above equation has the Arrhenius form
k
2
=Ae
-Ea/RT
AND
We therefore identify the activation energy with the
minimum kinetic energy along the line of approach that is
needed for a reaction and
That the pre-exponential factor A is a measure of the rate of
collisions occurring in gas
04/11/2012 03:26
04/11/2012 03:26
Collision rates in gases-The collision frequency z
NOTE: o= Collision cross-section =td
2
(Fig
below), and d = 1/2(d
A
+ d
B
)
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
13/2/2008
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
A Simple approach to calculate the collision frequency Z of the
molecules we consider the following hypothetical collision tube
Fig. 1
A: Freeze the positions of all molecules except one
B: Note what happen when the mobile molecule travels through the gas with
a mean speed C for a time At
C: Upon travelling, the molecule sweeps out a collision tube of cross-
sectional area o = td
2
and length CAt and thus volume = oCAt
d
d
d
d
d
d
<
<
<
>
>
Collision cross-section
miss
hit
hit
miss
hit
Fig 1
04/11/2012 03:26
The number of stationary molecules with centres
inside the collision tube is given by the volume
of the tube times the number density (N = N
A
[A] ) and
is NoC
rel
At = N
A
[A] oC
rel
At
- The collision frequency (the number of collisions per unit time) is
therefore N
A
[A] oC
rel
where [A] = the molar concentration of
molecule A, N
A
=
the Avogadros constant and C
rel
= is the relative
mean speed of the molecules
- For collision between different types of molecule, the
mean relative speed is
C
rel
= (8kT/t)
1/2
where = reduced mass = m
A
m
B
/m
A
+ m
B
-
Thus the collision frequency is
Z
AB
= o(8kT/t)
1/2
N
A
2
[A][B] and for similar molecules
the collision frequency is Z
AA
= o(8kT/t)
1/2
N
A
2
[A]
2
04/11/2012 03:26
The energy requirement
According to collision theory, the rate of change in [A]
molecules results from the collision density and the
probability that a collision occurs with sufficient energy
The energy requirement is incoporated by writing the
collision cross-section o as a function of the kinetic energy
of approach of the two colliding species
Also, the cross-section o(c) can be set equal to zero if the
kinetic energy of approach is below a certain threshold
value, c
a
Thus, d[A]/dt = -o(c)v
rel
N
A
[A][B]
04/11/2012 03:26
The relative KE, c
rel,
and the
speed are related by c
rel
= 1/2v
2
rel
gives the average Boltzmann disribution of energies and
hence the rate constant is recognized as
| |
dt
A d
= -{
( )
}
0
c o
u
rel
f(c)dc}N
A
[A][B]
2
k
= N
A
( )
}
0
c o
u
rel
f(c)dc
04/11/2012 03:26
Assuming that the reactive collision cross-section is zero
below c
a
, then we see that the variation of reactive cross-
section o(c) with energy is expressed by
( ) c o
= (
1
-
c
c
a
)
o
THUS k
2
= N
A
oC
rel
e
-Ea/RT
04/11/2012 03:26
The steric requirement: Consider the Arrhenius
parameters in the table below
Reaction A/(Lmol
-1
s
-1
)
experiment
A(Lmol
-1
s
-1
)
theory
Ea/
(kJmol
-1
)
Theory
exp
A
A
p =
2NOCl 2NO + 2Cl 9.4 X10
9
5.9 X 10
10
102.0 0.16
2ClO Cl
2
+ O
2
6.3 X 10
7
2.5 X 10
10
0.0 2.5 X 10
-3
H
2
+ C
2
H
4
C
2
H
6
1.24 X 10
6
7.4 X 10
11
180.0 1.7 X 10
-6
K + Br
2
KBr + Br 1.0 X 10
12
2.1 X 10
11
0.0 4.8
04/11/2012 03:26
The Arrhenius parameters data in the table above
show that :
There is a fair agreement between theory and
experiment
There are discrepancies
There are some cases where there are several orders
of magnitude smaller than experimental
It therefore shows that the collision energy is not the only
criterion for reaction and that some other feature such as
the relative orientation of the colliding species is important
NB: Larger than theory implies the reaction occurs more
quickly than the particles collide
04/11/2012 03:26
The disagreement between theory and experiment
is accommodated by introducing a steric factor, p,
and expressing the reactive cross-section, *
as a multiple of the cross-section, * = p =
Thus the rate constant becomes
k
2
= p(8kT/t)
1/2
N
A
e
-Ea/RT
Theory
exp
A
A
04/11/2012 03:26
Weaknesses of Collision theory (SCT):
No way to compute P from molecular
parameters
No way to compute c
a
from first principles.
Theory not quantitative or predictive.
Strengths of Collision theory (SCT):
Qualitatively consistent with observation
(Arrhenius equation).
Provides plausible connection between microscopic
molecular properties and macroscopic reaction rates.
Provides useful guide to upper limits for rate constant
k.
04/11/2012 03:26
SCT : a summary.
The major problem with SCT is that the threshold
energy c
a
is very difficult to evaluate from first
principles.
The predictions of the collision theory can be critically
evaluated by comparing the experimental pre-
exponential factor with that computed using SCT.
We define the steric factor P as the ratio between
the experimental and calculated A factors.
04/11/2012 03:26
We can incorporate P into the SCT expression for the
rate constant.
For many gas phase reactions P is considerably less
than unity.
Typically SCT will predict that Acalc will be in
the region 10
10
-10
11
L mol
-1
s
-1
regardless of
the chemical nature of the reactants and products.
What has gone wrong? The SCT assumption of hard
sphere collision neglects the important fact that
molecules possess an internal structure.
We need a better theory that takes molecular
structure into account. The activated complex theory
does just that .
04/11/2012 03:26
04/11/2012 03:26
Transition state theory (TST) = Activated complex
theory (ACT) = Absolute rate theory
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
20/2/2008
04/11/2012 03:26
RECAP: SCT HAS WEAKENESSES!
What has gone wrong? The SCT assumption of hard
sphere collision neglects the important fact that
molecules possess an internal structure.
We need a better theory that takes molecular
structure into account. The activated complex theory
(ACT) does just that .
04/11/2012 03:26
RECALL:
- An activated complex forms between reactants as they
collide and begin to assume the nuclear and electronic
characteristic of products
- We know that the change in potential energy associated
with formation of the activated complex accounts for the
activation energy of the reaction
- ACT considers a more detailed calculation of rate
constants using the concept of statistical thermodynamics
- ACT is an attempt to identify the principal features
governing the size of a rate constant in terms of a model
of the events that take place in the reaction
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
After the maximum, the potential energy falls as
the atoms rearrange in the cluster, and it reaches
a value characteristic of a product
Here the reactant molecules have come to such a
degree of closeness and distortion that a small
further distortion will result into the product
Note:This crucial configuration is called transition state of the
reaction and some molecules entering the transition state
might revert to reactants BUT if they pass through this
configuration then it is inevitable that products will be formed
from the encounter
NB: The activated complex is the area near the
potential maximum whereas the transition state
corresponds to the maximum itself
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
21/2/2008
04/11/2012 03:26
04/11/2012 03:26
Thermodynamic aspects of TST
- The concept of an equilibrium between reactants and the
activated complex enables us to treat activation processes in
terms of thermodynamic functions
- If we accept
K
in the Eyring equation as an equilibrium
constant, then we can express it in terms of a Gibbs energy of
activation,
G A
through the definition
RTInK G = A
or
p RTInK G = A
Where
G A
=
o
G
(activated complex) -
o
G
(reactants)
and K and
p K
are equilibrium constants
04/11/2012 03:26
Thus K=
p K
=
RT
G
e
A
and therefore from Eyring equation
Then the rate constant becomes
04/11/2012 03:26
04/11/2012 03:26
Now from the formal definition of activation energy,
( )
T
k
In RT E
a
c
c =
2
, then gives
RT H E
a
2 + A =
=
for reactions in gases
and
RT H E
a
+ A =
=
for reactions in solution (NOTE: All
thermodynamic quantities are in standard states)
HOW?
04/11/2012 03:26
So substituting H A in the rate equation we get
RT
E
R
S a
e e e
p
RT
h
kT
k
A
=
=
2
2
u
in gases
Whereas for reactions in solution, we get
RT
E
R
S a
e e e
p
RT
h
kT
k
A
=
=
1
2
u
in solution
04/11/2012 03:26
In general, in solution we have
RT
U
R
S
RT
H
R
S
e e
p
RT
h
kT
e e
p
RT
h
kT
k
= = = =
A
A A
A
~ =
u u
2
and in gas phase
RT
U
n
R
S
RT
H
R
S
e e e
p
RT
h
kT
e e
p
RT
h
kT
k
= = = =
A
A
A A
A
= =
u u
2
- Furthermore,
= = =
A + A = A V p U H
where for reactions occurring
in solution, the term
=
AV p
is quite small compared to
=
AU
and can therefore be neglected
- For the gas-phase reactions however, we have
n RT V p A = A
where
n A
is the change in moles from reactants to the
activated complex (NB: For unimolecular reaction
n A
= 0.)
For gas-phase reactions
= = =
A + A = A n RT U H
04/11/2012 03:26
When compared with the empirical Arrhenius equation
RT
E
a
Ae k
=
2 , we then see that in gases, the frequency factor
A =
R
S
e e
p
RT
h
kT
=
A
2
u
for gases
While for reactions in solution
A =
R
S
e e
p
RT
h
kT
=
A
u
What is the significance of
=
AS
and
=
AH ?
These equations enable us to interpret the probability factors
in terms of entropy of activation and thus
- The entropy of activation is negative when two reactant
species come to form one species
04/11/2012 03:26
- If reactants are only atoms or simple molecules, then
there is a relatively small amount of rearrangement of
energy among the various degrees of freedom in the
activated complex
- Consequently,
=
AS
will be either a small positive or
negative number, so that
R
S
e
=
A
and thus steric factor p is
close to unity. This implies that the reaction will proceed
much faster than predicted by the collision theory
- On the other hand, if complex molecules are involved in
a reaction,
=
AS
will be either a large positive or negative
number and this means a much slower rate will be
observed
04/11/2012 03:26
04/11/2012 03:26
It is useful to compare the expression of rate
constants discussed so far:
Empirical Arrhenius equation
Collision theory
TST
k
2
= p(8kT/t)
1/2
N
A
e
-Ea/RT
RT
E
R
S a
e e e
p
RT
h
kT
k
A
=
=
2
2
u
in gases
RT
E
R
S a
e e e
p
RT
h
kT
k
A
=
=
1
2
u
in solution
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
27/2/2008
04/11/2012 03:26
The pharmacokinetics of artensunate is that
following oral administtration .. reaches Cmax
within 45 to 90 minutes. The product is
effective against malaria by the same mechanism
of action.
04/11/2012 03:26
You may recall that thermodynamics can predict whether
or not a reaction will proceed but cannot tell us how fast
or slow will the reaction occur! NOW
Chemical kinetics deals with the understanding of
the factors that affect the rate of a chemical
reaction and elucidates information about the
mechanism of the reaction
(A reaction mechanism is a series of steps that defines the order
in which bonds are broken and new ones are formed, until the
final product is obtained)
chemical reaction kinetics is concerned with the
rates at which chemical reactions occur
NB: Generally, a rate is a change in some quantity wrt time
04/11/2012 03:26
Nature of reactants: Each reaction proceeds at its own rate.
Changing the nature of reactants will, in general, change the
rate of the reaction
The concentration of reactants: For a chemical reaction to
occur, the reactants must come close together and collide.
Because collisions are more frequent when concentrations
increase, then the reaction rates increase as the concentration
of the reactants increase
The temperature: In general, increasing the temperature
increases the rate of the reaction. The rate of most reactions
increases by a factor of 2 or 3 for a 10
o
C rise in temperature!
The nature of the solvent:Although not all reactions occur in
solution, but for those carried in solution, changing the solvent
will generally change the rate of the reaction
The presence of the catalyst
What factors affect the rate of a chemical reaction?
04/11/2012 03:26
Reactions we would like to speed up.
Drug delivery
Paint drying
Destruction of air pollutants in auto exhaust
Most industrial chemical processes
Breakdown of synthetic plastics in land fill sites
etc.
Reactions we would like to slow down.
Food decay (hence food additives)
Rubber decay
Human aging (antioxidants)
Fading of clothes (detergent additives)
Fires (fire retardents added to consumer products)
Ozone layer destruction (a big problem, CFCs etc)
Rusting of metallic objects etc
04/11/2012 03:26
To get the picture of rate in chemical kinetics let us
From the stoichiometry for the reaction it follows
that
| |
dt
D d
=
| |
dt
C
3
1
=
| |
dt
A d
=
| |
dt
B d
2
1
so there are several rates connected with the
reaction and as such , any one of them can
be considered the rate of the reaction!
04/11/2012 03:26
More elaboration on definition of rate
04/11/2012 03:26
04/11/2012 03:26
Rate law
04/11/2012 03:26
General rate expression: rate = v = k[A]
m
[B]
n
.
Where k = The rate constant of the reaction and is
independent of the concentrations of the reactants but
depends on temperature, the solvent (if in solution), the
particular reaction, and the catalyst (if any) that are present
in the reaction mixture
The exponents m, n, in the rate expression are small
integers (0,1,2,3,) or small fractions (1/2, 3/2, )
An experimentally determined equation of this kind is the rate law of the
reaction
A formal definition of a rate law: A rate law is an equation that expresses
the rate of reaction as a function of the concentrations of all the species
present in the overall chemical equation at some time i.e
v = f([A],[B], .)
04/11/2012 03:26
For homogeneous gas-phase reactions, it is often more
convenient to express the rate law in terms of partial pressures,
which are related to molar concentrations by
P
j =
RT[A] thus, v = f(P
A
, P
B
)
NB: The rate law of a reaction is determined
experimentally, and cannot in general be inferred
from the chemical equation for the reaction
What is the importance of a rate law in chemistry?
Once we know the rate law and the value of the
rate constant, we can:-
04/11/2012 03:26
Predict the rate of reaction from the composition of the
mixture
Predict the composition of the reaction mixture at a later
stage of the reaction
It serves as a guide to the mechanism of the reaction, for
any proposed mechanism must be consistent with the
observed rate law
04/11/2012 03:26
Units of rates and rate constant
The units of the rate of any reaction are
moles per liter per unit time, which may be
mol.L
-1
s
-1
.
The units the rate constant, k, however,
depend on the order of the reaction
04/11/2012 03:26
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
7/3/2006
04/11/2012 03:26
Determination of reaction order
Before anything can be said about the mechanism of a
reaction, the first task is to determine the order of the
reaction. This can be achieved by several methods such as
isolation, differential, half-life, integrated and relaxation
methods.
04/11/2012 03:26
Differential method (suggested by vant Hoff in 1884!)
Since rate =v = k[A]
n
, then by taking logarithms of
both sides, we obtain
logv = nlog[A] + logk
Then by measuring v at different concentration of A, we
can obtain the value of n from a plot of logv versus log[A]
log[A]
logV
slope = n
A plot of logv versus
log[A] gives a straight line
and the slope = order of
the reaction = n
04/11/2012 03:26
Integration method
Integration method is an obvious procedure whereby you
measure the concentration of the reactant(s) at various time
intervals of a reaction and substitute the data into integrated rate
equations
NOTE: Zero order reactions are relatively rare , among the best
studied reactions are those that occur on metal surfaces
04/11/2012 03:26
[A]
o
-[A]
Slope = k
Time
Plots for a zero order reaction
concentration of A
Rate
k
We see that for zero order reactions, the
rate of the reaction is independent of the
reactant concentration
04/11/2012 03:26
The expression [A] = [A]
o
e
-kt
shows that in a first-order reaction, the reactant
concentration decreases exponentially with time with a rate determined by k
The exponential decay of the reactant in
a first order reaction. The larger the rate
constant the more rapid the decay
04/11/2012 03:26
Plots for first order reaction
concentration of A
Rate
k
Slope = -k
Time
In[A]/[A]
o
04/11/2012 03:26
The first expression shows
that a plot of 1/[A] vs t
should give a straight line
with slope = k
The last expression lets us
predict the concentration of
A at any time after the start
of the reaction. It also shows
that the conc. of A
approaches zero more
slowly than in a first -order
reaction with the same initial
rate
04/11/2012 03:26
| |
dt
A d
= -k[A][B]
Note: The above rate law cannot be intergrated until we know how the concentration of B
is related to that of A.
Now consider the reaction A + B P where P denotes product and [A]
o
and [B]
o
are
initial concentrations for A and B respectively.
From the stoichiometry of the reaction, it follows that when the conc. of A has fallen to
[A]
o
X, the conc. of B will also have fallen to [B]
o
X because for each A that
disappears entails the same disappearance of one B. It follows that
| |
dt
A d
= -k([A]
o
x )([B]
o
- x) but because
| |
dt
A d
= -
dt
dx
, the rate law becomes
dt
dx
= k([A]
o
x )([B]
o
- x)
Thus
) ] )([ ] ([ x B x A
dx
o o
= kdt
04/11/2012 03:26
with the initial condition that x = 0 when t = 0 and so the integration of the rate law by
partial fractions gives
In
|
|
|
.
|
\
|
o
o
A
A
B
B
] [
] [
] [
] [
= ([B]
o
[A]
o
)kt
Half Lives
The half life of a substance (reaction) is the time taken for
the concentration of the reactant to fall to half its initial
concentration
This simple method is done by determining the dependence
of the half-life of a reaction on the initial concentration,
using the half life equation(s) from integrated rate laws.
04/11/2012 03:26
Half Lives
k
693 . 0
04/11/2012 03:26
- Unlike in the first order, the half-life of a substance in a
second-order reaction varies with the initial concentration
NOTE: A practical consequence of this dependence is that
species that decay by second-order reactions (sadly this
includes some environmentally unfriendly substances) may
persist in low concentration because their half-lives are long
when their concentrations are low!!.
04/11/2012 03:26
Relaxation methods
The term relaxation denotes the return of a system to
equilibrium
relaxation = return of a system to equilibrium
Relaxation methods are used to measure rate constants.
Apply a sudden perturbation to the equilibrium, e.g., a
temperature jump or a pressure jump, and monitor the
relaxation
Let us consider the response of a reaction rates to a sudden
change in temperature
Recall that the equilibrium composition of a reaction depends
on the temperature (provided A
r
H is nonzero), thus a shift in
temperature acts as a pertubation on the system [normally
done by LASER or microwave discharges)
04/11/2012 03:26
When a sudden temperature increase is applied to
a simple equilibrium that is first-order in
each direction it can be shown that the
composition relaxes exponentially to the new
equilibrium composition:
The exponential relation is
given by
x =
o
x
t
t
e
where t = k
a
+ k
b
and x is the departure from equilibrium at
new temperature and
o
x is the departure from equilibrium after
the temperature jump.
A
B
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
13/3/2006
04/11/2012 03:26
Reactions approaching equilibrium/Opposing reactions
The rate laws we have considered so far disregard the
possibility that the reverse reaction is important , and none of
the laws describes the overall rate when the reaction is close
to equilibrium
We know that most reactions are reversible to a certain extent,
so that we must consider both the forward and the reverse rate
At equilibrium, the products may be so abundant that
the reverse reaction must be considered. Thus for a
reversible reaction that proceeds by two elementary
steps:
A
B
k
1
k
-1
04/11/2012 03:26
We represent the net rate of change of A as
| |
dt
A d
= -k
1
[A] + k
-1
[B]
because when the concentration of A is reduced by the
forward reaction by a rate k[A] it is also increased by the
reverse reaction at a rate k
-1
[B]
Recall that at equilibrium, there is no net change in the concentration of A with time, that
is,
| |
dt
A d
= 0 so that -k
1
[A] + k
-1
[B] = 0
Thus k
1
[A] = k
-1
[B]
This leads to
| |
| | A
B
=
1
1
k
k
= K
Where K is the equilibrium constant for the reaction!
04/11/2012 03:26
NB: This equation is very important in chemistry
because it relates the thermodynamic quantity, the
equilibrium constant, K, and the quantities relating to
rates i.e If one of the rate constants can be measured,
then the other may be obtained if the equilibrium
constant is known.
For a more general reaction, the overall equilibrium constants for all the intermediate
stages of the reaction mechanism is
K =
1
1
k
k
x
2
2
k
k
x
Where the ks are the rate constants for the individual steps and the k
-
are for the
respective reverse steps
04/11/2012 03:26
The discussion of the relationship between reaction rates and
equilibria has its roots in a principle of great importance in
chemical kinetics and is called the principle of microscopic
reversibility!
The principle states that at equilibrium the rates of the
forward and reverse process are equal for every elementary
reaction occurring
This means that the process A B is exactly balanced by B A so
that the equilibrium cannot be maintained by a cyclic process, with the
reaction being A B and A C A on the opposite direction
The usefulness of this principle in chemical kinetics is that it tells us that
the reaction pathway for the reverse of the reaction is opposite of the
pathway for the forward reaction!
What does it mean when related to TST?
This means that the transition states (i.e the complex between the reactants
and products along the reaction pathway) for the forward and reverse reactions
are identical.
04/11/2012 03:26
Consecutive elementary reactions
Some reactions proceed through a formation of an
intermediate (I) (i.e species that occur in the reaction
mechanism but do not appear in the overall reaction
as in the consecutive unimolecular reactions
Let us suppose that initially only A is
present, and that its concentration is [A]
o
Remember that the first of the above rate
laws i.e
| |
dt
A d
= -k
a
[A] is a normal first order
and its intergrated rate law is [A] = [A]
o
t
k
a
e
04/11/2012 03:26
If we set [I]
o
= 0 the above differential
equation though is complex, it can be solved
to give the following solution:
[I] =
a b
a
k k
k
(e
-k
a
t
-e
-k
b
t
)[A]
o
At all times, [A] + [I] + [P] = [A]
o,
it
therefore follows that
[p] = {1 +
a b
t k
b
t k
a
k k
e k e k
a b
}[A]
o
When [A] is substituted in equation
dt
I d ] [
= k
a
[A] - k
b
[I], we get
dt
I d ] [
+ k
b
[I] = k
a
[A]
o
t
k
a
e
04/11/2012 03:26
If the three concentrations are plotted against time we
get the following figure
We see that:
Reactant A decays by
an ordinary first-order
intermediates rises to a
maximum at t
max
and
then falls to zero
The concentration of
the product P rises from
zero and reaches [A]
o
[p] = {1 +
a b
t k
b
t k
a
k k
e k e k
a b
}[A]
o
04/11/2012 03:26
04/11/2012 03:26
Generally, the rate determining step is the slowest step in a
mechanism and controls the overall rate of the reaction.
NOTE: The rate determining step is not just the slowest
step: it must be slow and crucial gateway for the formation of
products! If a faster reaction can also lead to products, then
the slowest step is irrelevant because the slow reaction can
then be side-stepped (see Fig below)
04/11/2012 03:26
The rate determining step (RDS)
04/11/2012 03:26
The rate law for a reaction with a rate determining step can
often be written down almost by inspection.
For instance, if the first step in the reaction mechanism is
rate determining step, then the rate of the overall reaction is
equal to the rate of the first step because all subsequent
steps are so fast that once the first intermediate is formed it
results immediately in the formation of products (Fig below).
04/11/2012 03:26
Pre-equilibrium
We have dealt with a mechanism of a simple consecutive
reaction. NOW
What happens if the intermediate reaches an equilibrium
with the reactants say A and B?:
This reaction involves pre-equilibrium, in which an
intermediate is in equilibrium with the reactants
NOTE: A pre-equilibrium arises when the rates of
formation of the intermediate and its decay back into
reactants are much faster than its rate of formation of
products, thus, the condition is possible when k'
a
>> k
b
but not when k
b
>> k'
a
Because we assume A, B and I are in equilibrium, then
K = [I]/[A][B], K = k
a
/k'
a
A + B
I P
k
a
k'
a
k
b
04/11/2012 03:26
The rate of formation of product P may therefore be
written d[P]/dt = k
b
[I], But [I] = K[A][B]
Thus d[P]/dt = k
b
K[A][B] =k[A][B]
where k = k
b
K = k
a
k
b
/k'
a
The above rate law has the form of a second-
order rate law with a composite rate constant.
NB: In writing these equations it is presumed
that the rate of reaction of I to form P is too
slow to affect the maintenance of the
equilibrium
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
14/3/2006
04/11/2012 03:26
The steady-state approximation
You may have noticed a considerable
increase in mathematical complexity as
soon as the mechanism has more than a
couple of steps
One approach to solving these complex
reactions is to integrate the rate laws
numerically and an alternative approach
is to make an approximation!
This approximation is termed steady-
state approximation and it assumes
that, after an initial induction period,
an interval during which the
concentrations of intermediates, I,
rise from zero, and during the major
part of the reaction, the rates of
change of concentrations of all
reaction intermediates are negligibly
small i.e ~ 0
dt
I d ] [
04/11/2012 03:26
For instance, we can apply the approximation to the
consecutive first-order mechanism.
RECALL THAT
Then [I] ~ ka [A] /kb
upon substituting this value on the above equation d[P]/dt
= kb[I] we get d[P]/dt = kb[I] ~ ka[A]!! We therefore see that P
is formed by a first order decay of A, with a rate constant ka,
the rate constant of the slower, RDS.
For SSA we set d[I]/dt = 0 such that k
a
[A] - k
b
[I] ~ 0
[P] ~ (1 - e-kat)[A]o showing that the formation of the final
product P depends on only the smaller of the two rate constants!
i.e the rate of formation of P depends on the rate at which I is formed,
not on the rate at which I changes into product P
04/11/2012 03:26
1. First identify the intermediates (i.e species that occur in the
reaction but do not appear in the overall reaction)
2. Write expressions for their net rates of formation
3. Set all net rates of change of the concentrations of
intermediates approximately equal to zero
4. Solve the resulting equations algebraically.
How the rate law is determined using Steady-State
Approximation?
04/11/2012 03:26
Homework: Use the steady-state approximation to devise the rate law
for the decomposition of N
2
O
5,
on the basis of the following mechanism.
N
2
O
5 NO
2
+ NO
3
k
a
k'
a
NO
2
+ NO
3
NO
2
+ O
2
+NO
k
b
NO + N
2
O
5
NO
2
+ NO
2
+ NO
2
k
c
4NO
2
(g) + O
2
(g)
N
2
O
5
(g)
04/11/2012 03:26
04/11/2012 03:26
O
N
R
N
O
R
+
2
4a R = H
4b R = NO
2
4c R = OCH
3
5a R = H
5b R = NO
2
5c R = OCH
3
Catalytic reactions consist of a reaction cycle formed by a
series of elementary reaction steps for example DA reaction
04/11/2012 03:26
Mechanism of Lewis acid catalysed DA in water
Cu
2+
O
N
O
2
N
O
N
O
2
N
Cu
2+
N
O
NO
2
Cu
2+
N
O
NO
2
04/11/2012 03:26
Rate expression are indispensable in the application of
catalysed reactions, in the design of a chemical reactors, and
their process control
The rate expression for a catalysed reaction is in general a
function of many parameters i.e v = f(catalyst, T, k
i
, P
i
,
K
i
..Keq)
Catalysts are conveniently placed into two groups
viz. Homogeneous and Heterogeneous
Homogeneous catalysts have the same phase as the
reactants
Heterogeneous catalysts: The catalyst has a different
phase from the reaction mixture.
Rates of catalysed reactions and groups of catalysts:
04/11/2012 03:26
Heterogeneous catalysts provide surface upon
which reactants bind; this binding facilitates
encounters between reactants and hence increase
the rate of the reaction
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
20/3/2006
04/11/2012 03:26
Enzyme kinetics
One of the most fascinating studies in chemical kinetics is the
investigation of enzyme catalysis
An enzyme-catalysed reaction is usually characterised by a
very large increase in the rate (on the order of 10
6
to 10
12
)
and high specificity
Specificity means that the enzyme molecule is capable of
selectively catalysing certain reactants, the substrate, while
discriminating against
An enzyme usually contains one or more ACTIVE SITE(S)
where reactions with substrate take place
An active site may contain only a few amino acid residue,
the rest of the protein molecule is however required to
maintain the 3-D integrity of the network
WHY ENZYMES ARE SPECIFIC?
04/11/2012 03:26
In 1890s, Fischer proposed that enzyme specificity may be
explained in terms of Lock-and-Key theory. In this theory,
the active site is assumed to have a rigid structure,
similar to a lock whereas a substrate molecule has the
complimentary structure and function like a key
NOTE: This theory has been modified to take into account the
flexibility of proteins in solution and to explain the
phenomenon of cooperativity
04/11/2012 03:26
In enzymology, we mostly study the basic mathematical
treatment of enzyme kinetics
It is almost a custom to study enzyme kinetics by involving
the measurement of initial rate (v
o
) of a reaction. Why??
It minimises the rate of reverse reaction, since the rate increases
with the concentration of the product
During the course of the reaction, there may be appreciable heat
and /or pH changes which may alter the conformation of the
enzyme and hence affect the rate of the reaction
In some cases, the product formed may bind to the enzyme in
such a way it inhibits its function
The initial rate corresponds to a known fixed substrate
concentration
In enzyme kinetics we assume that the enzyme-substrate
binding ratio is 1:1 and when the reaction profile is followed,
the substrate concentration drops (Fig. next slide)
04/11/2012 03:26
The rate increase rapidly at low concentrations of the
substrate but then gradually levels off at high
concentrations of substrate
Mathematical analysis show that
the relationship between the
initial rate u
o
and [S] can be
defined in terms of the equation
of a rectangular hyperbola
=
o
v
| |
| | S b
S a
+
where a and b are constants
How do we explain the dependence of the initial rate on
the concentration of a substrate?
04/11/2012 03:26
Michaelis-Menten kinetics (1913)
Michaelis and Menten proposed a theory to explain the
dependence of the initial rate on concentration
They considered the following scheme:(Compare with pre-
equilibria!)
A + B
I P
k
a
k'
a
k
b
E is the enzyme, S is the "substrate" (the molecule on
which the enzyme does its work), and ES is an
enzyme-substrate complex. (It is presumed that the
substrate must somehow bind to the enzyme before
the enzyme can do its work.)
VS
04/11/2012 03:26
The initial rate of product formation v
o
is given by
v
o =
The concentrations of the enzyme and substrate at a time
shortly after the start of the reaction(initial time) are
[E] = [E]
o
- [ES] and [S] = [S]
o
- [ES]
However experimental conditions are usually such that
[S]
o
>> [E]
o
so that [S]
o
>> [ES] or [S] ~ [S]
o
Michaelis and Menten assumed that k
-1
>> k
2
so that the first
step (i.e the formation ES complex) can be treated as a rapid
equilibrium process AND therefore the dissociation constant
is given by
| || |
| |
| | | | ( )| |
| | ES
S ES E
ES
S E
k
k
K
o
s
= = =
1
1
04/11/2012 03:26
Solving for [ES], we obtain
Substituting [ES] in the rate equation
we get
Note: a = k
2
[E]
o
and b = K
s
The physical significance of a and b may be understood as
follows: At high substrate concentration i.e [S] >> b so that
At these conditions all the enzyme molecules are in the complex
form, so that the initial rate must be at its maximum value (v
max)
| | | |
| | S K
S E
ES
s
o
+
= ] [
| | | |
| | S K
S E k
dt
P d
s
o
o
+
= =
2
] [
v compare with
| |
| | S b
S a
+
| |
| |
| |
| |
| |
o o
E k
S
S a
S b
S a
2
= ~
+
= v
04/11/2012 03:26
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
21/3/2006
04/11/2012 03:26
If you postulate a short time after the enzyme and the substrate are mixed,
the concentration of the enzyme-substrate will reach a constant value can
you apply SSA? Yes you can! (Briggs and Halden did that in 1925)
| |
| || | | |
| || | | | ] [ 0
] [
2 1 1
2 1 1
ES k ES k S E k
ES k ES k S E k
dt
ES d
=
=
but
| | | | ] [ES E E
o
=
| | | | ( )| | | | | | ES k ES k S ES E k
o 2 1 1
0 =
| | ( )| | ( )| |
| | | | | | ( )| | ES k k S ES k S E k
ES k k S ES E k
o
o
2 1 1 1
2 1 1
] [
] [
+ + =
+ =
04/11/2012 03:26
04/11/2012 03:26
If we compare
| | | |
| |
M
o
K S
S E k
+
2
with
| | | |
| | S K
S E k
s
o
+
2
we see that both equations have
similar dependence on substrate concentration, however
s M
K K =
unless
1
k
>>
2
k
because
1
2 1
k
k k
K
M
+
=
~
1
1
k
k
In this treatment we also define the maximum rate
max
v exactly the same as before such
that
max
v = | |
o
E k
2
and hence the above equation can also be written as
| |
| |
M
o
K S
S
+
=
max
v
v
In principle, this shows that both
max
v
and M
K
can be
determined from a plot of
o
v
versus [S] (see Fig.below)
1/2 v
max
v
max
K
M
Initial rate (v
o
)
[S]
| |
| | S b
S a
+
04/11/2012 03:26
In practice, it is not very useful determining the v
max
since it is
difficult to determine the asymptotic value v
max
at a very high
substrate concentrations
A more satisfactory approach is by employing the
Lineweaver-Burk approach where you take a double
reciprocal of equation
where you get
| |
| |
M
o
K S
S
+
=
max
v
v
| |
max max
M
1
K
u
+
u
=
u S
1
o
where
| |
line straight a gives
S
1
Versus
o
1
u
04/11/2012 03:26
From the plot,
M
K
and max
v
can be obtained from the
slope and intercepts of the straight line.
04/11/2012 03:26
What is the significance of
max
v
,
2
k
and
M
K
??
- The maximum velocity max
v
represents the maximum rate
attainable, that is, it is the rate when the total enzyme concentration
is present as the enzyme-substrate complex!
- The kinetic constant
2
k
(in units of s
-1
) in the equation max
v
=
| |
o
E k
2
is called the turnover number(also k
cat
). The turnover number of an
enzyme is the number of substrate molecules converted into
product per unit time, when the enzyme is fully saturated with the
substrate! NOTE: The TONs of most enzymes for their
physiological substrates fall in the range of 1 to 10
5
s
-1
.
-
M
K lacks a simple interpretation.
However when
1
k >>
2
k then
M
K can be
equated to the dissociation constant K
s
When this condition is met,
M
K becomes
a measure of the strength of the ES
complex
04/11/2012 03:26
- A large
M
K
indicates weak binding and
small
M
K
indicate strong binding
- Generally,
M
K
(in units of M) is
customarily reported together with
2
k
and
max
v
for enzyme-catalysed reactions.
This is because
M
K
depends on pH,
temperature, substrate etc., and thus, its
value serves to characterise a particular
enzyme-substrate system under certain
specified conditions.[In most cases
M
K
lies between 10
-2
and 10
-7
M).
Enzyme inhibition
04/11/2012 03:26
Enzyme catalysed reactions are prone to inhibition by
molecules that interfere with the formation of the product
Actually, many drugs for the treatment of diseases are
designed to function by inhibiting enzymes!
NOTE: This is one of the strategy towards developing a
drug to treat AIDS. In this case, it involves a steady
administration of a specially designed protease
inhibitor I.e the drug inhibits an enzyme that is key to
the formation of protein envelope surrounding the
genetic material of the human immunodeficiency virus
(HIV). Without a properly formed envelope, HIV can not
replicate in the host organism.
Inhibitors are compounds(negative catalysts) that decrease the rate of an enzyme-
catalysed reaction
04/11/2012 03:26
Enzyme inhibition
There are two types of enzyme inhibitions: Reversible and irreversible
Enzyme Inhibition
Reversible Irreversible
Competitive Noncompetitive Uncompetitive
04/11/2012 03:26
Competitive Inhibition
In this case the substrate S and the inhibitor I compete
for the same active site. The reactions are:
E + S
E S P + E
E + I E I
where the complex EI does not form products
The situation can be treated by Michaelis-Menten kinetics. Applying the steady-state
approximation for ES, we obtain
| |
| |
(
|
|
.
|
\
|
+
|
|
.
|
\
|
+
=
1
max
1 1
K
I
S
K
M
o
v
v where
| || |
| | EI
I E
K =
1
The Lineweaver-Burk equation is given by
| |
| |
max 1 max
1 1
1
1
v v v
+
|
|
.
|
\
|
+ =
S K
I K
M
o
Thus a straight line is obtained by plotting
o
v
1
against
| | S
1
at constant | | I
04/11/2012 03:26
END
END
END
04/11/2012 03:26
CH344
27/3/2006
04/11/2012 03:26
Competitive Inhibition
In this case the substrate S and the inhibitor I compete
for the same active site. The reactions are:
E + S
E S P + E
E + I E I
where the complex EI does not form products
The situation can be treated by Michaelis-Menten kinetics. Applying the steady-state
approximation for ES, we obtain
| |
| |
(
|
|
.
|
\
|
+
|
|
.
|
\
|
+
=
1
max
1 1
K
I
S
K
M
o
v
v where
| || |
| | EI
I E
K =
1
The Lineweaver-Burk equation is given by
| |
| |
max 1 max
1 1
1
1
v v v
+
|
|
.
|
\
|
+ =
S K
I K
M
o
Thus a straight line is obtained by plotting
o
v
1
against
| | S
1
at constant | | I
04/11/2012 03:26
04/11/2012 03:26
Dividing equation
| |
| |
M
o
K S
S
+
=
max
v
v
by
| |
| |
(
|
|
.
|
\
|
+
|
|
.
|
\
|
+
=
1
max
1 1
) (
K
I
S
K
M
inhibition o
v
v
we
get
( )inhibition
o
o
v
v
=
| |
| |
I M I
M
K K K S
I K
+
+ 1
NOTE: To overcome this competitive inhibition, we need to increase
the substrate concentration relative to the inhibitor; that is, at high
substrate concentration, [S]K
I
>> K
M
K
I
and
( )inhibition
o
o
v
v
~
| |
| |
I
M
K S
I K
+ 1
~ 1
04/11/2012 03:26
Noncompetitive inhibition
The Lineweaver-Burk equation is given by
| |
| |
|
|
.
|
\
|
+ + =
I
M
o
K
I
V S V
K
1
1 1 1
max max
v
- A noncompetitive inhibitor usually does not bind at the active site
of the enzyme. The reactions are:
E + S
E S P + E
E + I E I
ES + I ESI
- Both EI and ESI do not form products. Noncompetitive inhibition
can not be reversed by increasing the substrate concentration.
- The initial rate is given by
| |
| | ( )
| |
| || |
| |
| || |
| | ESI
I ES
EI
I E
K
K
I
S K
S V
I
I
M
o
= =
(
|
.
|
\
|
+ +
= where ,
1
max
v
,
o
v
1
VS | | S
1
gives a straight line
Noncompetitive Inhibition
04/11/2012 03:26
04/11/2012 03:26
Dividing
| |
| |
M
o
K S
S
+
=
max
v
v
by
| |
| | ( )
| |
(
|
.
|
\
|
+ +
=
I
M
o
K
I
S K
S V
1
max
v
we get
( )inhibition
o
o
v
v
= 1 +
| |
I
K
I
- NOTE: The results above confirms that the degree of
noncompetitive inhibition is independent of [S] and depends only
on [I] and K
I
04/11/2012 03:26
UNCOMPETITIVE NHIBITION
- An uncompetitive inhibitor does not bind to the enzyme; instead ,
it binds reversibly to the enzyme-substrate complex to yield an
inactive ESI complex. The reactions are:
E + S
E S
P + E
ES + I ESI
- The ESI does not form a product. Again, since I does not interfere
with the formation of ES, uncompetitive inhibition cannot be
reversed by increasing the substrate concentration. The initial rate is
given by
o
v
| |
| |
| |
|
.
|
\
|
+ +
=
I
M
K
I
S K
S V
1
max
where
| || |
| | ESI
I ES
K
I
=
04/11/2012 03:26
The Lineweaver-Burk equation is given by
o
v
1
=
| |
| |
|
|
.
|
\
|
+ +
I
M
K
I
V S V
K
1
1 1
max max
A straight line is obtained by plotting
o
v
1
versus
| | S
1
at constant
| | I
Dividing
| |
| |
M
o
K S
S
+
=
max
v
v
by
=
o
v
1
| |
| |
|
|
.
|
\
|
+ +
I
M
K
I
V S V
K
1
1 1
max max
we get
( )inhibition
o
o
v
v
=
| |
| |
| |
M
I
M
K S
K
I
S K
+
|
.
|
\
|
+ + 1
If conditions are such that [S] >> K
M
, then the equation becomes
( )inhibition
o
o
v
v
=
| |
| |
| | S
K
I
S K
I
M
|
.
|
\
|
+ + 1
= 1 +
| |
I
K
I
04/11/2012 03:26
04/11/2012 03:26
The catalytic efficiency of Enzymes
The catalytic efficiency, c, of an enzyme is the ratio of
Now recall that and
Thus
M
cat
K
k
1
2 1
k
k k
K
M
+
=
v
max =
k
2
[E]
o
M
cat
K
k
= c =
| |
2 1
2 1
2 1
1 max
k k
k k
k k
k
X
E
V
o
+
=
+
The higher the value of c, the more efficient is the enzyme (catalyst).
NOTE: Because k
1
is the rate constant for the formation of a complex
from two species that are diffusing freely in solution, the maximum
efficiency is related to the maximum rate of diffusion of E and S in
solution. This happens when k
2
>> k
-1
!
04/11/2012 03:26
04/11/2012 03:26
END
END
END
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
Conductance and Conductivity
The nature of molecular motion in solution can be obtained
by studying the motion of ions in solution (electrolytes).
By studying the transport of charge through electrolyte
solutions, it is possible to get a picture of how the events occur
and the results can be extrapolated to uncharged (neutral)
species
- An electrolyte is an ionic conductor i.e solution, a liquid or a
solid.
- Electrolytes dissociate in aqueous solution and the ions
formed act, to a large extent, as free, independent entities (Cf
kinetic-molecular theory of ideal gases)
- The ions experience van der Waals forces, interactions of the
ions between themselves and interaction of ions with the
solvent
04/11/2012 03:26
- The study of this motion of ions is done by measuring the electrical
resistance R, of the solution.
- The inverse of this resistance,
R
1
is the conductance, G, of the
solution (units
1
O mho or SI unit siemens, S) i.e
R
G
1
=
- Conductance is proportional to the cross-sectional area and is
inversely proportional to length
- That is
l
A
G
and therefore
l
A
G k =
where
ty conductivi = k
Or
= = G
A
l
k
Cell constant x G
- With the conductance in siemens and the dimensions in metres; it
follows that the SI unit of conductivity (
k
) are siemens per metre
(Sm
-1
)
04/11/2012 03:26
- The conductivity of the solution depends on the number of ions
present in the solution and we normally introduce the molar
conductivity, m
A
, which is defined as
c
m
k
= A
where c = the molar concentration of the added electrolyte
- The concentration dependence of molar conductivities show that
there are two classes of electrolytes i.e Strong and weak
electrolytes
- The characteristics of a strong electrolyte is that its molar
conductivity depends only slightly on the molar concentration
(generally decreases slightly as the concentration is increased)
- The characteristic of weak electrolyte on the other hand is that its
molar conductivity is normal at concentrations close to zero, but
falls sharply to low values as the concentration increases (Fig
overleaf)
NOTE: The classification is solvent dependent (a solute could be a
weak electrolyte in one solvent but strong in another)
04/11/2012 03:26
04/11/2012 03:26
STRONG ELECTROLYTES:
- Strong electrolytes are substances that are virtually fully ionized in
solution, and include ionic solids and strong acids.
- Due to this complete ionization, the concentration of ions in
solution is proportional to the concentration of strong electrolyte
added
- At low concentration the molar conductivities
m
A
of strong
electrolytes vary linearly with the square root of the concentration:
c
o
m m
k A = A
This variation is called Kohlrauschs law.
- The constant
o
m
A
is the limiting molar conductivity, the molar
conductivity in the limit of zero concentration (when the ions
are effectively infinitely far apart and do not interact with one
another).
- The constant
k
is found to depend more on the stoichiometry
of the electrolyte than on its specific identity
04/11/2012 03:26
- Kohlrauschs also showed that
o
m
A
can be expressed as the sum of
contributions from its individual ions
- Let us denote the limiting molar conductivity of cations by
+
and that of anions by
then Kohlrausch law of
independent migration of ions states that
+ +
+ = A v v
o
m where
+
v v and
are the numbers of cations and anions per formula unit of
electrolyte (e.g
1 = =
+
v v
for HCl or NaCl where
, 1 =
+
v 2 =
v
for
MgCl
2
)
WEAK ELECTROLYTES
- Weak electrolytes are not fully ionized in solution.
- The marked concentration dependence of their molar conductivities
arises from the displacement of the equilibrium towards products at
low molar concentrations.
HA
+
H
3
O+
+
A
(aq)
H
2
O (l)
(aq)
(aq)
04/11/2012 03:26
HA
A
O H
a
a
a a
K
+
=
3
- The conductivity depends on the number of ions in solution, and
therefore on the degree of ionization,
o
, of the electrolyte; and
for weak acids, we talk of the degree of deprotonation.
- For the acid HA at a molar concentration c, at equilibrium
[H
3
O
+
] = oc, [A
-
] = oc, [HA] = (1- o)c
- Ignoring activity coefficients, the acidity constant K
a
, is
approximately
o
o
=
1
2
c
K
a
from which it follows that
|
|
.
|
\
|
+ = 1
4
1
2
2
1
a
a
K
c
c
K
o
04/11/2012 03:26
- The acid is fully protonated at infinite dilution, and its molar
conductivity is then
o
m
A
. Because only a fraction o is actually
present as ions in the actual solution, the measured molar
conductivity
o
m m
A = A o
with o given above.
- By knowing the K
a
, we can use it to predict the concentration
dependence of the molar conductivity
- We can also use the concentration dependence of m
A
in
measurements of the limiting molar conductance by rearranging
equation
o
o
=
1
2
c
K
a
into
a
K
c o
o
+ = 1
1
and by using
o
m m
A = A o
we
obtain
( )
2
1 1
o
m a
m
o
m m K
c
A
A
+
A
=
A
(Ostwalds dilution law)
04/11/2012 03:26
This shows that a plot of
m
A
1
against c m
A
, then the intercept at c = 0
will be
o
m
A
1
as shown in the figure below.
( )
2
1 1
o
m a
m
o
m m
K
c
A
A
+
A
=
A
04/11/2012 03:26
END
END
END
04/11/2012 03:26
04/11/2012 03:26
THE MOBILITIES OF IONS
Why ions move at different speed? Why they have different molar
conductivities and why the molar conductivities of strong
electrolytes decrease with the square root of the molar concentration?
We shall see these when we study the drift speed, the ion mobility
and the transport numbers!
The drift speed:
- When the potential difference between two electrodes a distance
l apart is |, the ions in the solution between them experience a
uniform electric field of magnitude E where
l
| A
= E
In this field, an ion charge ze experiences a force of magnitude
l
ze
zeE F
| A
= =
04/11/2012 03:26
NOW: A cation responds to application of the field by accelerating
to the negative electrode and the anion responds by accelerating to
the positive electrode
- However, as the ion moves through the solvent, it experiences a
frictional retarding force, F
fric,
proportional to its speed.
- Assume Stokes formula for a sphere of radius a and speed s
applies at microscopic scale, then we can write this retarding
force as
F
fric
= fs, where f = 6qta and s = speed
- The two forces act in opposite directions, and the ions quickly
reach a terminal speed, the drift speed when the accelerating
force is balanced by the viscous drag.
- The net force is zero when
f
zeE
s =
- It follows that the drift speed of an ion is proportional to the
strength of the applied field. We write
uE s =
where u is called the mobility of the ion
04/11/2012 03:26
Recall that
1 ........ .......... ..........
6
zeE
f
zeE
a
s
tq
= =
......2 .......... uE........ = s
Comparing equation 1 and 2 we get
a tq 6
ze
f
ze
u Mobility, = =
Ionic mobilities in water at 298 K, u/(10
-8
m
2
s
-1
V
-1
)
H
+
36.23
Na
+
5.19
K
+
7.62
Zn
2+
5.47
OH
-
20.64
Cl
-
7.91
Br- 8.09
SO
2-
4
8.29
Why proton has a very high molar conductivity?
04/11/2012 03:26
- The proton, although it is very small, has a very high molar conductivity. Both
proton and
17
O-NMR show that the times characteristic of protons hopping
from one molecule to the next are about 1.5 ps, which is comparable to the time
that neutron scattering shows it takes a water molecule to reorient through
about 1 rad. According to the Grotthuss mechanism, there is an effective
motion of a proton that involves the rearrangement of bonds in a group of water
molecules.
- However, the actual mechanism is still highly debatable. Attention now focuses
on the H
9
O
4
+
unit in which the nearly trigonal planar H
3
O
+
ion is linked to three
strongly solvating H
2
O molecules. This cluster of atoms is itself hydrated, but
the hydrogen bonds in the secondary sphere are weaker than in the primary
sphere. It is envisaged that the rate-determining step is the cleavage of one of
the weaker hydrogen bonds of this secondary sphere (Fig. a).
04/11/2012 03:26
- After this bond cleavage has taken place, and the released
molecule has rotated through a few degrees (a process that takes
about 1 ps), there is a rapid adjustment of bond lengths and
angles in the remaining cluster, to form an H
5
O
2
+
cation of
structureH
2
O H
+
OH
2
(Fig. b).
03/04/2006 5:27
04/11/2012 03:26
- Shortly after this reorganization has occurred, a new H
9
O
4
+
cluster forms as other molecules rotate into a position where they
can become members of a secondary hydration sphere, but now
the positive charge is located one molecule to the right of its
initial location (Fig. c).
03/04/2006 5:30
- According to this model, there is no coordinated motion of a
proton along a chain of molecules, simply a very rapid hopping
between neighbouring sites, with a low activation energy. The
model is consistent with the observation that the molar
conductivity of protons increases as the pressure is raised, for
increasing pressure ruptures the hydrogen bonds in water.
04/11/2012 03:26
- NOW again, because the drift speed governs the rate at which
the charge is transported, we expect the conductivity to decrease
with increasing solution viscosity and ion size.
- Experiments however confirm the prediction on size is true for
bulky ions (e.g R
4
N
+
,
RCO
-
2
) but not for small ions! Why?
- This can be explained when we notice that the radius a in the
Stokes formula is the effective radius in the solution taking into
account all the water molecules it carries in its hydration sphere
NOW
- Because small ions give rise to stronger electric fields than large
ones, it means small ions are more extensively solvated than big
ones (Note: The electric field at the surface of a sphere of radius r
is proportional to
2
r
ze
)
- Thus, an ion of small ionic radius may have a large hydrodynamic
radius a,because it drags more solvent through the solution as it
migrates!
04/11/2012 03:26
Mobility and conductivities
- Mobility gives us a link between measurable and theoretical
quantities
- The relation between an ions mobility and its molar conductivity
is given by
zuF =
Where F = The Faraday constant (F= N
A
e)
- Thus, for the solution itself in the limit of zero concentration
(i.e when there are no interionic interactions)
(recall that
+ +
+ = A v v
o
m
) then, the law of independent
migration of ions becomes
( )F u z u z
o
m + + +
+ = A v v
- For a symmetrical z:z electrolyte, such as CuSO
4
with z = 2, the
above equation simplifies to
( )F u u z
o
m +
+ = A
04/11/2012 03:26
04/11/2012 03:26
Transport numbers/Transference
- The transport number (transference),
t
, is defined as the
fraction of total current carried by the ions of a specified type.
- Now, for a solution of two kinds of ions, the transport numbers
of the cations
( )
+
t
and anions
( )
t
are
I
I
t
=
- where
I
is the current carried by the ions and I is the total
current through the solution
- Because the total current is the sum of the cation and anion
currents, it follows that
1
1
= +
+
t t
- The limiting transport number,
o
t
, defined as the fraction
of the total current carried by the ions of specified type
for the limit of zero concentration of the electrolyte
solution
04/11/2012 03:26
- Note that the current that can be ascribed to each type of ion is
related to the mobility of the ion by
cFAE zu
l
cFA zu
I v =
| A v
=
where E = Electric charge
z = charge
c v
= molar concentration of ion
F = Faraday constant
A = Cross-sectional area
l = length
| A
=Potential difference
- Hence the relationship between limiting transport number
o
t
and mobility
u
is
+ + +
+
=
u z u z
u z
t
o
v v
v
- However, because + +
v z
=
v z
for any electrolyte, the
above equation becomes
+
+
=
u u
u
t
o
04/11/2012 03:26
- Moreover, because the ionic conductivities are related to
the mobilities by
zuF =
, therefore it follows that:
+ +
+
=
v v
v
o
t
But recall that + +
+ = A v v
o
m and thus
o
m
o
t
A
=
+
=
+ +
v
v v
v
- For each type of ion
= A v
o
m
o
t
NB:As a result, because there are independent ways of measuring
transport numbers of ions, we can determine the individual ionic
conductivities and the ionic mobilities
Measurement of transport numbers
04/11/2012 03:26
Measurement of transport numbers
- Transport numbers can be measured by methods such as Moving
boundary method or Hittorf method.
- In the Moving Boundary Method, the motion of a boundary
between two ionic solutions having a common ion is observed as
the current flows.
- Let MX (leading solution) be a salt of interest and NX
(indicator solution) a salt giving denser solution. The indicator
solution (NX) occupies the lower part of the vertical tube of
cross-section area A (Fig overleaf). The leading solution (MX)
occupies the upper part of the tube. There is a sharp boundary
between the two solutions. The indicator solution must be
denser than the leading solution, and the mobility of M ions
must be greater than that of N ions. Therefore, if any M ions
diffuse into the lower solution, they will be pulled upward
more rapidly than the N ions around them, and the boundary
will reform.
04/11/2012 03:26
The interpretation of the experiment
makes use of the relationship between
the distance, l, moved by the
boundary in the time interval t for
which a current I is passed.
t I
clAF z
t
A
=
+
Hence, by measuring the distance
moved, the transport number and
hence the conductivity and mobility
of the ions can be determined!
04/11/2012 03:26
Conductivities and ion-ion interactions
How do we account why the molar conductivities of
stronger electrolytes decrease with the square root of the
molar concentration? Recall
c
o
m m
k A = A
This can qualitatively be explained in terms of the
properties of the ionic atmosphere around each ion (Fig)
04/11/2012 03:26
The ionic atmosphere based on Debye-Huckel theory in that there is
a tendency for anions to be found around cations and of cations to be
found around anions (cicled to emphasize). Note: The ions are in
ceaseless motion, and the figure represents a snapshot of their
motion.
Ionic atmosphere causes (1) relaxation effect (2) Electrophoretic
effect
- To accommodate the effect of motion, the picture of an ionic
atmosphere looks as a spherical haze of charge
04/11/2012 03:26
- Now because the ions forming the atmosphere do not
adjust to the moving ion immediately, the atmosphere is
incompletely formed in front of the moving ion and
incompletely decayed behind the ion(Fig)
(a)In the absence of an electric field,
the ionic atmosphere is spherically
symmetric, BUT (b) when a field
is present it is distorted and the
centres of negative and positive
charge no longer coincide!)
04/11/2012 03:26
- The overall effect is the displacement of the centre of charge of
the atmosphere a short distance behind the moving ion
- Because the two charges are opposite, the result is a retardation of
the moving ion and this reduction of the ions is what we call the
relaxation effect
- Another effect on the ionic atmosphere in the motion of ions is
that because moving ions experience a viscous drag; the drag is
enhanced because the ionic atmosphere moves in the opposite
direction to the central ion
- The enhanced viscous drag is called the electrophoretic effect
and reduces the mobility of the ions, and hence also reduces their
conductivities AND
It can therefore be shown that the decrease in molar conductivity
due to these two effects is proportional to the square root of
concentration!!
04/11/2012 03:26
- A quantitative formulation is rather complex. Debye-Huckel-
Onsgager theory attempts to obtain the quantitative
expression.
- The theory leads to a Kohlrausch-like expression in which
k = A + B
o
m
A
with
2
1
2 2
2
3
|
.
|
\
|
=
RT
eF z
A
c tq
2
1
3
2
24
|
.
|
\
|
=
RT RT
F qz
B
c tc
where c = electric permittivity of the solvent
q = 0.586 for 1,1-electrolyte
NOTE: The slopes of the conductivity curves are predicted to depend
on the charge type of the electrolyte, in accord with Kohlausch law,
and comparisons between theory and experiment agree quite good at
very low concentration (0.001 M) depending on the charge type (Fig)
04/04/2004 20:47
04/11/2012 03:26
04/11/2012 03:26
Ionic atmosphere causes (1) relaxation effect (2) Electrophoretic
effect
- To accommodate the effect of motion, the picture of an ionic
atmosphere looks as a spherical haze of charge
04/11/2012 03:26
- Many macromolecules, such as DNA, are charged and move
in response to an electric field . This motion is called
electrophoresis. Electrophoretic mobility is a result of a
constant drift speed, s, reached by an ion when the driving
force zeE (where, as usual, ze is the net charge and E is the
field strength) is matched by the frictional force fs.
a
s
tq
= =
6
zeE
f
zeE
- Therefore, the mobility of a macromolecule in an electric
field depends on its net charge, size (and hence molar
mass), and shape. The latter two factors are implied by the
dependence of s on f. in the above equation.
04/11/2012 03:26
- Electrophoresis is a very valuable tool in the separation of
biopolymers from complex mixtures, such as those resulting
from fractionation of biological cells. We shall consider three
techniques commonly used: viz gel elctrophoresis, capillary
electrophoresis and isoelectric focusing. They differ in the
manner in which the drift speeds of biomolecules are
controlled in order to achieve separation of a mixture into its
components.
Gel Electrophoresis
- In gel electrophoresis, migration takes place through a gel slab. A
common gel material for the study of proteins is cross-linked
polyacrylamide. In most cases, the goal of the experiment is to
separate a sample according to the molar masses of its
components. However, the drift speed formula, states that the
shape and charge will also determine the drift speed. For example,
proteins with the same size but different net charge travel along the
slab at different speeds.
04/11/2012 03:26
- Moreover, most proteins bind a constant amount of ion, so
that the net charge per protein is well regulated. Under these
conditions, different proteins in a mixture may be separated
according to size only. The molar mass of each constituent
protein is estimated by comparing its mobility in its rod-like
complexes form with a standard sample of known molar
mass. However, molar masses obtained by this method are
not as accurate as those obtained by other techniques, such as
Matrix-Assisted Laser Desorption/Ionization Time Of Flight
(MALDI-TOF) and size exclusion microscopy.
- One way to avoid this problem and to effect a separation by
molar mass is to denature the proteins in a controlled way.
Sodium dodecyl sulphate is an anionic detergent that is very
useful in this respect: it denatures proteins, whatever their
initial shape, into rods by forming a complex with them.
04/11/2012 03:26
Capillary Electrophoresis
- The drift speed attained by polymers in traditional
electrophoresis methods are rather low; as a result, several
hours are often necessary to effect good separation of
complex mixtures.
- According to drift speed formula, one way to increase the
drift speed is to increase the electric field strength. However,
there are limits to this strategy because very large electric
fields can heat the large surfaces of an electrophoresis
apparatus unevenly, leading to a non-uniform distribution of
electrophoretic mobilities and poor separation.
a
s
tq
= =
6
zeE
f
zeE
04/11/2012 03:26
- In capillary electrophoresis, the sample is dispersed in a
medium (such as methylcellulose) and held in a thin glass or
plastic tube with diameters ranging from 20 to 100 m. The
small size of the apparatus makes it easy to dissipate heat
when large electric fields are applied. Excellent separation
may be effected in minutes rather than hours.
Each polymer fraction emerging from the capillary can be
characterized further by other techniques, such as MALDI-TOF.
Isoelectric Focusing
- Naturally occurring macromolecules acquire a charge when
dispersed in water. An important feature of proteins and other
biopolymers is that their overall charge depends on the pH of
the medium.
04/11/2012 03:26
- For instance, in acidic environments protons attach to basic
groups and the net charge is positive; in basic media the net
charge is negative as a result of proton loss. At the isoelectric
point, the pH is such that there is no net charge on the
biopolymer. Consequently, the drift speed of a biopolymer
depends on the pH of the medium, with s = 0 at the
isoelectric point.
- Isoelectric focusing is an electrophoresis method that
exploits the change of drift speed with pH. Consider a
mixture of distinct proteins dispersed in a medium with a pH
gradient along the direction of an applied electric field. Each
protein in the mixture will stop moving at a position in the
gradient where the pH is equal to the isoelctric point. In this
manner, the protein mixture can be separated into its
components.
04/11/2012 03:26
END
END
END
04/11/2012 03:26
04/11/2012 03:26
- Strong electrolytes though completely dissociate into ions in the
solid state, do not behave as though the ions are independent
particles in aqueous solutions.
- You may recall from chemical equilibrium topic that the
equilibrium expression is not strictly constant if concentrations are
used as the measure of active masses of the reacting species.
RECAP
- As you may remember, the physical properties of salt solutions,
such as conductivity and freezing point, suggest that the ions may
be clustered together, with positive ions having more negative
than positive ions in their immediate neighbourhood, and negative
ions in turn having an excess of positive ions around them (IONIC
ATMOSPHERE).
- This time-averaged, spherical haze around the central ion, in which
counter ions outnumber ions of the same charge as the central ion, has a
net charge equal in magnitude but opposite in sign to that on the cenral
ion, and is called its ionic atmosphere
04/11/2012 03:26
- In order to obtain agreement between experimental and
theoretical equilibrium, the chemist multiplies actual
concentrations (e.g. molalities, molarities) by certain
numbers, called activity coefficients
, to obtain effective
concentrations, called activities a.
- The activity of a species A is defined as follows: a =
[A].
From this definition, it can be seen that the more ideally a
solution behaves, the closer the activity coefficient to unity.
Indeed, at infinite dilution,
=1 and a = [A].
Ion activities:
- Under such conditions, the effectiveness of ions in
determining the rate of chemical reactions, and also in
altering physical properties of the solvent, is less than it
would be were each ion capable of acting independently.
Only in very dilute solutions are ions sufficiently free of the
influence of neighboring ions to act as independent
particles.
04/11/2012 03:26
- The activity a is a kind of effective mole fraction of the
solution and is related to the molality, b by
u
=
b
b
a
where
activity coefficient,
, depends on the composition,
molality, and temperature of the solution.
- As the solution approaches ideality (in that it obeys Henrys
law) at low molalities, the activity coefficient tends towards
1.
1
and u
b
b
a
as
0 b
Alternatively,
- There is an enormous amount of experimental evidence to
indicate that electrostatic between ions and between ions and
solvent molecules can cause large differences between activity
and concentration. Such interactions are general, not just
between ions undergoing chemical reaction.
04/11/2012 03:26
- For example, many compounds show increased solubility in the
presence of electrolytes containing no ions which react chemically
with ions of the precipitate! The solubility product constant, K
sp
, of
BaSO
4
, for instance, increases from 1.0 x 10
-10
in water to 2.9 x 10
-
10
in 0.010 M aqueous KNO
3
at 298 K. Clustering of NO
-
3
ions
about Ba
2+
and of K
+
ions about SO
2-
4
tends to shield Ba
2+
and SO
2-
4
ions from each other and to hamper their effectiveness in forming
BaSO
4
- The term used to measure the magnitude of the electrostatic
environment of a solute is called ionic strength I, and this
will treated in the next lecture.
|
.
|
\
|
=
u
b
b
z
i
i
i
2
1
I
Where is the charge number of an ion (positive for cations and
negative for anions) bi is its molality
i
z
04/11/2012 03:26
Ion activities at standard states:
- Because interactions between ions are so strong, the
approximation of replacing activities by molalities is valid only
in very dilute solutions (infact less than 10
-3
mol kg
-1
in total ion
concentration)
- Recall that the chemical potential of a solute in a real solution at
any molality is related to its activity a by
a ln RT + =
u
- where the standard state is a hypothetical solution with molality,
u
b
= 1mol kg
-1
in which the ions are behaving ideally.
Because all the deviations from ideality are carried in the activity
coefficient, the chemical potential can be written
u
ln ln RT
ideal
RT RTInb + = + + = where
ideal
is the
chemical potential of the ideal-dilute solution of the same molality
04/11/2012 03:26
Note: The geometric mean of activity coefficient of monovalent
solution is
( )
2
1
+
=
Generally, for a salt with ions like M
n+
X
m-
, the geometric mean
activity is
m n
m n
+
+
=
|
.
|
\
|
/ 1
04/11/2012 03:26
- The long range and strength of the Coulombic interaction
between ions means that it is likely to be primarily responsible for
the departure from ideality in ionic solution and to dominate all
other contributions to nonideality
- This possible domination is the basis of the Debye-H ckel theory
of ionic solutions (Peter Debye and Erich Hckel 1923)
- A qualitative account of the theory is given
- Oppositely charged ions attract one another. As a result, anions
are likely to be found near cations in solution, and vice versa
- NB: Overall, the solution is electrically neutral, but near any
given ion there is an axcess of counter ions i.e Averaged over
time, counter ions are more likely to be found near any given ion
The Debye-Huckel Limiting law
04/11/2012 03:26
END
END
END
04/11/2012 03:26
04/11/2012 03:26
- The long range and strength of the Coulombic interaction
between ions means that it is likely to be primarily responsible for
the departure from ideality in ionic solution and to dominate all
other contributions to nonideality
- This possible domination is the basis of the Debye-Hckel theory
of ionic solutions (Peter Debye and Erich Hckel 1923)
- A qualitative account of the theory is given
- Oppositely charged ions attract one another. As a result, anions
are likely to be found near cations in solution, and vice versa
- NB: Overall, the solution is electrically neutral, but near any
given ion there is an axcess of counter ions i.e Averaged over
time, counter ions are more likely to be found near any given ion
The Debye-Huckel Limiting law
04/11/2012 03:26
DH theory is a quantitative theory of electrolyte solution which
is based on a rather simple model and allows us to calculate the
quantity of activity coefficient
from the basic properties of
the solution.
The mathematical details of DHs treatment are too difficult
to present here rather a simple underlying assumptions will
be presented.
Debye and Huckel began by assuming the following:
1. Electrolytes are completely dissociated into ions in
solution
2. The solutions are dilute, that is, the concentration is
0.01 m or lower
3. On the average each ion is surrounded by ions of
opposite charge , forming the ionic atmosphere
The Debye-Huckel Limiting law
04/11/2012 03:26
Working from these assumptions, Debye and Huckel calculated the
average electric potential at each ion caused by the presence of other
ions in the ionic atmosphere.
The Gibbs free energy of the ions was then related to the
activity coefficient of the individual ion.
Since neither +
nor
could be measured directly, the final
result is expressed in terms of the mean activity coefficient of
the electrolyte as follows
( )
I z z log
2
3
+
c
=
T
10 x 1.824
6
The Debye-Huckel Limiting law
04/11/2012 03:26
- The model leads to the result that at very low concentrations the
activity coefficient can be calculated from the Debye-Hckel
limiting law
2
1
AI z z 509 . 0 log
+
=
where A= 0.509 for an aqueous solution at 298 K and I is the
dimensionless ionic strength of the electrolyte solution:
I =
|
.
|
\
|
u
b
b
z
i
i
i
2
1
In this expression
i
z
is the charge number of an ion (positive for
cations and negative for anions) b
i
is its molality
Since most studies are carried out in water at 298 K (we have
c
= 78.54 and T = 298 K) and the above equation
becomes.simplified as written below:
The Debye-Huckel Limiting law
04/11/2012 03:26
- The model leads to the result that at very low concentrations the
activity coefficient can be calculated from the Debye-Hckel
limiting law
2
1
log AI z z
+
=
where A= 0.509 for an aqueous solution at 298 K and I is the
dimensionless ionic strength of the solution:
I =
|
.
|
\
|
u
b
b
z
i
i
i
2
1
In this expression
i
z
is the charge number of an ion (positive for
cations and negative for anions) b
i
is its molality
04/11/2012 03:26
- It is important to realise that the sum extends over all the ions
present in the solution! For instance, a solution consisting two
types of ion of molalities +
b
and
b
( )
u
b z b z b I /
2
1
2 2
+ +
+ =
Note: The ionic strength emphasizes the charges of the ions because
the charge numbers occur as their squares!
04/11/2012 03:26
APPLICATION OF DH THEORY
The DH limiting law can be applied to study the solubility of
proteins. How soluble the protein is in the aqueous solution
depends on the temperature, pH, dielectric constant, ionic
strength, and other characteristics of the medium.
We shall only be concerned with the influence of ionic strength.
Suppose we want to investigate the effect of ionic strength on the
solubility of AgCl. The dissolution process is
AgCl(s)
Ag+ (aq) + Cl (aq)
The thermodynamic solubility product for the process,
o
sp
K
is
-
Cl Ag
o
sp
a a K
+
=
+ +
=
Cl Ag
-
Cl Ag
o
sp
b b K
04/11/2012 03:26
Because, activity, a = molality x activity coefficient!
-
Cl Ag
o
sp
b b
K But
+
+
=
=
sp
sp
Cl Ag
K because
K
where sp
K
= the apparent solubility product!
Thus the thermodynamic solubility product represents the true
value of the solubility product, which in general is different
from the apparent solubility product.
Since
=
+
2
Cl Ag
Then we can write
sp
o
2
sp
2
o
K
K
K K
s
s
or = =
04/11/2012 03:26
Taking the logarithm of both sides of the above equation and
rearranging, we obtain
I z log
2
1
o
sp K
+
=
|
|
.
|
\
|
=
z 0.509
K
log
sp
which comes to the DH limiting law!
For a 1:1 electrolyte, the solubility product can directly be
related to the solubility S itself because
( ) S = 2
1
sp
K
and
( )
2
1
sp
K
o o
S =
Where S and
o
S are apparent and thermodynamic solubilities
in moles per liter.
Hence
I z
+
= z 0.509
S
S
log
o
04/11/2012 03:26
The solubility of solute may increase or decrease with the
increase of ionic strength.
The increase in solubility, caused by the increase in ionic
strength, is called SALTING-IN EFFECT whereas the decrease
in solubility with increasing ionic strength of the solution is
called the SALTING-OUT EFFECT.
04/11/2012 03:26
Extended Debye-Huckle limiting law
- The simple DH equation, or limiting law, has been found to
give useful results only in dilute solutions. As the ionic
strength increases, mean activity coefficients calculated from
the equation are significantly smaller than the experimental
value.
- For example, the calculated value for the mean activity
coefficient in 0.10 M HCl is 0.69, whereas the experimental
value is 0.8!
- Debye and Huckel later extended their theoretical treatment,
assuming that ions are not point charges, but have significant
size, and that they are present in solutions of greater ionic
strength than originally assumed. They derived the so called
extended Debye-Huckel equation,
04/11/2012 03:26
I 1
I Z
log
B
B
0.512Z
A
o +
=
where
o
is the diameter of the ion in angstrom (1 = 10
-10
m), and B
a term which depends on the absolute temperature and the dielectric
constant of the solvent.
- Its value at 298 K using the dielectric constant for pure water is
0.328. The term
o
, sometimes called the ion-size parameter, is
said to correspond to the distance of closest approach of the
cation and anion.
Note: Activity coefficients calculated by the extended DH
equation are in good agreement with experimentally measured
mean values up to ionic strengths of about 0.1. For solution of
higher ionic strength, a number of modifications of the DH
equation have been proposed.
04/11/2012 03:26
- The energy, and therefore the chemical potential, of any given
central ion is lowered as a result of its electrostatic interaction
with its ionic atmosphere.
- This lowering of energy appears as the difference between the
molar Gibbs energy G
m
and the ideal value
ideal
m
G
of the solute,
and hence can be identified with RT
ln
.
NB:The stabilization of ions by their interaction with their ionic
atmospheres is part of the explanation as to why chemists
commonly use dilute solutions, in which the stabilization is less
important, to achieve precipitation of ions from electrolyte
solutions. In other words, as the solution becomes diluted, the
solute becomes increasingly stabilised and the practical
consequence of this is that it is very difficult to remove traces of a
solute from such a solution!
04/11/2012 03:26
Example: Q. Calculate the mean activity coefficient
, of 5.0
x10
-3
molkg
-1
KCl(aq) at 298 K.
Ans: We start by writing
2
1
log AI z z
+
=
But
( )
u
b b b I /
2
1
+
+ =
=
u
b
b
Where b is the molality of the solution (note b
-
= b
+
= b) and
therefore we have
2
1
2
1
509 . 0 509 . 0 log
|
.
|
\
|
= =
b
b
I
( )
2
1
3
2
1
10 5 509 . 0 509 . 0 log
= = x I
log
= -0.036
Hence
= 0.92
04/11/2012 03:26
END
END
END
04/11/2012 03:26
Lecture on 2 May 2006
04/11/2012 03:26
Electrochemistry
Electrochemistry is the science which deals with
the consequences of the transfer of electric charge
from one phase to another.
Electron transfer occurs at interfaces between a
metallic conductor (an electrode) and an ionic
conductor (an electrolyte).
An electrochemical reaction is a heterogeneous
process which involves electron transfer across a
phase boundary or interface.
Electrochemical reactions are labelled as redox
(Reduction / oxidation) processes.
Oxidation is the loss of electrons (LEO) and
Reduction is the gain of electrons (GER).
04/11/2012 03:26
Why do we study electrochemistry?
Every battery is an electrochemical cell and indeed the
measure of its voltage is a Gibbs free energy indicator.
In nerve impulses in biological systems there are
electrochemical potentials
Concentration gradients across cell and other membranes
cause electrochemical potential differences.
In manufacturing and chemical etching, electrochemical
reactions abound.
Photosynthesis, where carbon is fixed in living matter is a basic
electrochemical process
The transport of oxygen in blood by hemeogloblin and the
change of the oxidation states of iron involves electrochemistry
Therefore:A basic understanding of the principles behind
electrochemical processes to understand all these processes is
needed.
04/11/2012 03:26
04/11/2012 03:26
Know
The
Differen
ces
Betwe
en
These!
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
Basic Concepts
04/11/2012 03:26
END
END
END
04/11/2012 03:26
Lecture on 8 May 2006
04/11/2012 03:26
- Electrochemical cells are grouped based on the general
thermodynamic nature of the reaction (i.e expressed as
whether the change in Gibbs energy is -ve or +ve)
- A galvanic cell uses a spontaneous reaction inside it (-G ) to
generate electrical energy) i.e The reacting system does work
on the surrounding and all batteries are voltaic!
- An electrolytic cell on the other hand uses electrical energy to
drive a non-spontaneous reaction (+G) i.e The surroundings
do work on the reacting system
- It is important to note that all electrochemical cells have
several common features:Have two electrodes (Anode and
Cathode) and the oxidation half-reaction takes place at the
anode and the reduction half-reaction takes place at the
cathode
An overview of electrochemical cells (RECAP):
04/11/2012 03:26
04/11/2012 03:26
04/11/2012 03:26
Shorthand nomenclature
Rather than writing out the full chemical
equation, we use a shorthand notation:
Zn(s) | Zn
2+
(aq) || Cu
2+
(aq) | Cu(s)
Zn(s) + Cu
2+
(aq) Zn
2+
(aq) + Cu(s)
Cathode half-cell Anode half-cell
Salt bridge
Phase boundary
Phase boundary
Electrons flow this way
04/11/2012 03:26
Cell reaction
- To write the cell reaction corresponding to a cell diagram,
we first write the RH half reaction as a reduction
Right hand electrode:
Cu
2+
+
2e
-
(aq)
Cu(s)
Left hand electrode: Zn
2+
(aq) + 2e
-
Zn(s)
.....1
.....2
Hence the overall cell reaction is the difference i.e eqn 1 - eqn 2 resulting into
Cu
2+
(aq)
+ Zn(s)
Cu(s) Zn
2+
+
(aq)
Thus in the cell Zn(s)|ZnSO
4
(aq)||CuSO
4
(aq)|Cu(s) above, the
two electrodes and their reduction half-reactions are:
- The current produced by a galvanic cell arises from the
spontaneous chemical reaction taking place inside it
04/11/2012 03:26
THE CELL POTENTIAL
- A cell in which the overall cell reaction has not reached
chemical equilibrium can do electrical work as the reaction
drives electrons through an external circuit
- The work that a given transfer of electrons can accomplish
depends on the potential difference between the electrodes
The potential difference is called the cell potential and is
measured in volts, V
When the cell potential (potential difference) is large, a given
number of electrons travelling between the electrodes can do a
large amount of electrical work and vice versa
A cell in which the overall reaction is at equilibrium can do no
work, and the cell potential is zero
The resulting potential difference when the cell (not the cell
reaction) is at equilibrium is what is called the electromotive
force (emf), E, of the cell
04/11/2012 03:26
- From thermodynamics, the maximum electrical work that a system
(the cell) can do is given by the value of G,
Welec., max
= G
(RECALL: The reaction Gibbs energy, rG, is defined as the slope of
the graph of the Gibbs energy plotted against the extent of reaction)
THE CELL POTENTIAL
Note: F is the Faraday constant, F = eN
A
,
the magnitude of the charge
per mole of electrons
.
This equation is the key connection between
electrical measurements on the one hand and thermodynamic
properties on the other.
04/11/2012 03:26
- The equation -
v
FE = rG, forms a key connection between
electrical energy on the one hand and the thermodynamic properties
on the other i.e
i) By knowing the reaction Gibbs energy at a specified
composition, we can state the cell emf at that composition
ii)The -G corresponds to a spontaneous cell reaction and to a +ve
emf
iii) The driving power of a cell (i.e its emf) is proportional to the
slope of the Gibbs energy with respect to the extent of reaction
iv) It is plausible that a reaction that is far from equilibrium (when
the slope is steep) has a strong tendency to drive electrons through an
external circuit and when the slope is close to zero (when the cell
reaction is close to equilibrium) the emf is small
04/11/2012 03:26
Nernst equation:
- We can extend the relation of the emf to the activities of the
participants in the cell reaction
(Recall: , where Q is the reaction quotient)
NOW
- From the Nernst equation, we see that the standard emf of a
cell can be interpreted as the emf when the reactants and
products are in their standard states, for then all activities are
1, so Q = 1 and InQ = 0
04/11/2012 03:26
Lecture on 9 May 2006
04/11/2012 03:26
- A concentration cell is a galvanic cell in which the
electrode materials and the solutions in both half-cells are
composed of the same substances; only the
concentrations of the two solutions differ
- Consider a concentration cell where the solution M has L
and R molalities
- ).
04/11/2012 03:26
- Because a cell cannot drive a
current through a circuit when the two electrode
compartments are identical! (i.e rG = 0 ).
04/11/2012 03:26
- We know that a galvanic cell is a combination of two
electrodes , and each one can be considered as making a
characteristic contribution to the overall cell potential
- The specially selected electrode is the standard hydrogen
electrode (SHE) at all temperatures:
Pt(s)|H
2
(g)|H
+
(aq)
- The standard potential, (
u
E ), of another couple is assigned by
constructing a cell in which it is RH electrode and the standard
hydrogen electrode is the LH electrode.
- For instance, the standard potential of the Ag
+
/Ag couple is the
standard emf of the following cell
Pt(s)|H
2
(g)|H
+
(aq)||Ag
+
(aq) |Ag(s), (
u
E ) = (Ag+/Ag) = +0.80 V
- Because it is impossible to measure the contribution of a single
electrode, we define the potential of one electrode as zero and
then assign values to others on that basis
04/11/2012 03:26
The standard emf of a cell can be obtained EITHER
OR
- From the difference of the standard potentials of electrodes i.e
upon subtracting the standard potential of the LH electrode
(cathode) from the standard potential of the RH electrode
(anode)
NOTE
THUS
04/11/2012 03:26
04/11/2012 03:26
Applications of Standard Potentials:
- Cell emfs are a convenient source of data on the Gibbs
energies, enthalpies and entropies of reactions
NOTE:
- Electrochemical series
i.e The metallic elements (and hydrogen)
arranged in order of their reducing power
as measured by their standard potentials
in aqueous solution
04/11/2012 03:26
04/11/2012 03:26
i.e
2
RT
H
dT
dInK
r
o
A
=
or
R
rH
T
d
dInK
o
A
=
)
1
(
- After combining the results we obtain the standard
reaction enthalpy to be
|
|
.
|
\
|
= A + A = A
dT
dE
T E F rS T rG rH
o
o o o o
v
04/11/2012 03:26
- Thus, an electrochemical technique is used to obtain
standard reaction entropies and through them entropies of ions
in solution
- It is interesting to note that the above expressions provide a
noncalorimetric method for measuring
o
H
r
A
and, through the
convention,
( ) 0 , = A
+
aq H fH
o
, the standard enthalpies of formation
of ions in solution
- This means electrical measurements can be used to calculate
all thermodynamic properties!!!
04/11/2012 03:26
Course Outline (2 lectures/w and 2Tutorial/w)
Microscopic theories of reaction rates: Collision and
transition state theories
Rates and mechanisms of chemical reactions (reactions
rates and equilibria, Elementary reactions and enzyme
catalysed reactions)
Electrolytes in solutions
The Debye-Hckel theory of interionic interaction and
ionic activity coefficients
Electromotive force and electrochemical cells
CH 344: CHEMICAL KINETICS AND
ELECTROCHEMISTRY (2 units)
Dr. E.B. Mubofu
Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- Practical Class Two Matlab PracticalДокумент6 страницPractical Class Two Matlab PracticalPradyumnaSadgirОценок пока нет
- EnzymeKinetics PDFДокумент21 страницаEnzymeKinetics PDFaathiraОценок пока нет
- Biochemistry Ch. 8 EnzymesДокумент9 страницBiochemistry Ch. 8 EnzymesBryantОценок пока нет
- Mindray Bs 200Документ13 страницMindray Bs 200Roberto AriasОценок пока нет
- Transient State Kinetic Measurements 1Документ3 страницыTransient State Kinetic Measurements 1OLUWASEGUN K AfolabiОценок пока нет
- Exam Iii: Only V VДокумент8 страницExam Iii: Only V Vss shinodaОценок пока нет
- Journal of Biological Engineering: Tinkercell: Modular Cad Tool For Synthetic BiologyДокумент17 страницJournal of Biological Engineering: Tinkercell: Modular Cad Tool For Synthetic Biologyr4ging8ullОценок пока нет
- Enzyme KineticsДокумент72 страницыEnzyme Kineticsitokki otoya100% (1)
- Derivation of Reaction Rate Expression With Equilibrium AssumptionДокумент6 страницDerivation of Reaction Rate Expression With Equilibrium AssumptionViaTS13Оценок пока нет
- Amylase Enzyme AssayДокумент7 страницAmylase Enzyme AssayBoby Chrisol Joseph100% (1)
- Lecture Notes-Growth Kinetics - Growth PhasesДокумент24 страницыLecture Notes-Growth Kinetics - Growth PhasesNhan Nguyen100% (1)
- CHE 46: Biochemical EngineeringДокумент30 страницCHE 46: Biochemical EngineeringJunn Edgar LibotОценок пока нет
- 2021 ATNAA Competitve Product InhibitionДокумент14 страниц2021 ATNAA Competitve Product InhibitionvinhОценок пока нет
- Bluff EnzymesДокумент51 страницаBluff EnzymesJay Andrea Vea Dayuday-IsraelОценок пока нет
- Kinetic Model of Thermophilic - Lactate Fermentation by Bacillus Coagulans Combined With Real-Time PCR QuantificationДокумент9 страницKinetic Model of Thermophilic - Lactate Fermentation by Bacillus Coagulans Combined With Real-Time PCR QuantificationJohanaОценок пока нет
- One-Pot Synthesis of Protein-Embedded Metal Organic Frameworks With Enhanced Biological ActivitiesДокумент5 страницOne-Pot Synthesis of Protein-Embedded Metal Organic Frameworks With Enhanced Biological ActivitiesnathaloaОценок пока нет
- Food EnzymologyДокумент1 052 страницыFood Enzymologychemistryreactions100% (4)
- Enzyme KineticsДокумент10 страницEnzyme KineticsQuenneBelocuraОценок пока нет
- 09931275D L265 UV Lab Software Users GuideДокумент127 страниц09931275D L265 UV Lab Software Users GuideAbdul KalimОценок пока нет
- A Kinetic Study of The Enzymatic Hydrolysis of Cassava StarchДокумент6 страницA Kinetic Study of The Enzymatic Hydrolysis of Cassava StarchInternational Journal of Science and Engineering Investigations100% (2)
- International Journal of Pharma and Bio Sciences: Subtilis Dj5 Into Gelatin Film by Glutaraldehyde CrosslinkingДокумент10 страницInternational Journal of Pharma and Bio Sciences: Subtilis Dj5 Into Gelatin Film by Glutaraldehyde CrosslinkingAnggaAnugrah LiberoeAdjhaОценок пока нет
- M Tech BiopharmaceuticalДокумент38 страницM Tech BiopharmaceuticalarunbaskaranjОценок пока нет
- Syllabus 3102 Spring 2018Документ5 страницSyllabus 3102 Spring 2018Arthur LembongОценок пока нет
- Palaganas Hw2 Che 197Документ5 страницPalaganas Hw2 Che 197Elah Palaganas100% (1)
- Biochem Lec Transes (Week 4)Документ6 страницBiochem Lec Transes (Week 4)Juren LasagaОценок пока нет
- Biokinetics PDFДокумент28 страницBiokinetics PDFWazif ZakwanОценок пока нет
- 20.320 Exam 2 Review Problem Solutions April 20, 2006 1. No InhibitionДокумент3 страницы20.320 Exam 2 Review Problem Solutions April 20, 2006 1. No InhibitionMovie Scene BankОценок пока нет
- Different Enzyme Kinetic Models: Eleanore Seibert and Timothy S. TracyДокумент13 страницDifferent Enzyme Kinetic Models: Eleanore Seibert and Timothy S. TracyValeria CazaresОценок пока нет
- Chapter 2. ExerciseДокумент5 страницChapter 2. Exercisedhuy2399Оценок пока нет
- Mathematical Modeling of Biological Processes: Avner Friedman Chiu-Yen KaoДокумент152 страницыMathematical Modeling of Biological Processes: Avner Friedman Chiu-Yen KaoSamuel YufraОценок пока нет