Вам также может понравиться
- Overview of LymphomaДокумент42 страницыOverview of Lymphomaadamu mohammadОценок пока нет
- Chronic Lymphocytic LeukemiaОт EverandChronic Lymphocytic LeukemiaMichael HallekОценок пока нет
- Non Hodgkin LymphomaДокумент9 страницNon Hodgkin LymphomaAhmad SaifulОценок пока нет
- Lymphoma HerfindalДокумент56 страницLymphoma HerfindalAanshi ShahОценок пока нет
- Case StudyДокумент10 страницCase Studysabrown109Оценок пока нет
- LymphomaДокумент69 страницLymphomaDawit g/kidanОценок пока нет
- Lymphoma Shofik EDITED BY SAYAM 8.53PMДокумент35 страницLymphoma Shofik EDITED BY SAYAM 8.53PMRAGHU NATH KARMAKERОценок пока нет
- Leukemia Research Paper Thesis StatementДокумент9 страницLeukemia Research Paper Thesis Statementaparnaharrisonstamford100% (1)
- Non - Hodgkin LymphomaДокумент25 страницNon - Hodgkin LymphomaRo RyОценок пока нет
- Pediatric Non-Hodgkin Lymphoma - Background, Etiology, EpidemiologyДокумент11 страницPediatric Non-Hodgkin Lymphoma - Background, Etiology, EpidemiologytopОценок пока нет
- Acute Leukemia: Thirunavukkarasu MurugappanДокумент22 страницыAcute Leukemia: Thirunavukkarasu MurugappanFelix Allen100% (1)
- Acute Leukemia HerfindalДокумент35 страницAcute Leukemia HerfindalAanshi ShahОценок пока нет
- 1) Explain The Risk Factors For CLL, Including The Relevance of Monoclonal B Cell Lymphocytosis (MBL)Документ6 страниц1) Explain The Risk Factors For CLL, Including The Relevance of Monoclonal B Cell Lymphocytosis (MBL)Fahim MahmudОценок пока нет
- Pediatrik LeukemiasДокумент14 страницPediatrik LeukemiasLefrina GusrianiiОценок пока нет
- Non-Hodgkin's LymphomasДокумент23 страницыNon-Hodgkin's LymphomasshevmyrОценок пока нет
- Acute Myeloid Leukemia-BookДокумент15 страницAcute Myeloid Leukemia-BookRhoda Lom-ocОценок пока нет
- Acute Leukemia RodaksДокумент14 страницAcute Leukemia RodaksLoiLoiChanОценок пока нет
- Nk-Cell Lymphomas of The Head and Neck: AuthorДокумент8 страницNk-Cell Lymphomas of The Head and Neck: AuthorCharmila SariОценок пока нет
- Medicine Seminar Combined-1Документ30 страницMedicine Seminar Combined-1Deepanshu KumarОценок пока нет
- The Role of Immunophenotyping in The Diagnosis of Acute Leukemia. A Narrative Literature ReviewДокумент6 страницThe Role of Immunophenotyping in The Diagnosis of Acute Leukemia. A Narrative Literature ReviewmyhalfsmileОценок пока нет
- Acute Lymphoblastic LeukemiaДокумент80 страницAcute Lymphoblastic LeukemiaGizo YitayewОценок пока нет
- Childhood LeukemiaДокумент10 страницChildhood LeukemiafrancyriveraОценок пока нет
- Non-Hodgkin's Lymphoma: A Histopathologic and Prognostic EvaluationДокумент12 страницNon-Hodgkin's Lymphoma: A Histopathologic and Prognostic Evaluationchimbimb100% (2)
- Hodgkein and Nonhodgkein LymphomaДокумент57 страницHodgkein and Nonhodgkein Lymphomasamar yousif mohamedОценок пока нет
- Hutter2010 LeucemiaДокумент10 страницHutter2010 LeucemiasarabisimonaОценок пока нет
- Acuteleukemia: Hayley Rose-Inman,, Damon KuehlДокумент18 страницAcuteleukemia: Hayley Rose-Inman,, Damon KuehlJorge Tovar AvilaОценок пока нет
- L-3 Introduction To LeukemiaДокумент26 страницL-3 Introduction To LeukemiaAbood dot netОценок пока нет
- Wall 2016Документ15 страницWall 2016Jose AbadiaОценок пока нет
- Background: Pediatric Acute Lymphoblastic LeukemiaДокумент3 страницыBackground: Pediatric Acute Lymphoblastic LeukemiaNitin KumarОценок пока нет
- Acute Lymphoblastic LeukemiaДокумент30 страницAcute Lymphoblastic LeukemiaHarancang Kahayana0% (1)
- PATHO LEC WBC Lymph Nodes Spleen Thymus Part1 CompressedДокумент84 страницыPATHO LEC WBC Lymph Nodes Spleen Thymus Part1 CompressedAngelo HinonОценок пока нет
- Shan Shal 2012Документ15 страницShan Shal 2012Jose AbadiaОценок пока нет
- Leukemia Myeloproliferation, Myelodysplasia: Lita Septina Peyakit Dalam FK UMSUДокумент47 страницLeukemia Myeloproliferation, Myelodysplasia: Lita Septina Peyakit Dalam FK UMSUJr SparkОценок пока нет
- Blockxiv Neoplasms Lymphoid 2006Документ54 страницыBlockxiv Neoplasms Lymphoid 2006Ryo RyozОценок пока нет
- Childhood Malignancies - LeukaemiasДокумент4 страницыChildhood Malignancies - Leukaemiasvictortayor_26105009Оценок пока нет
- The Qualitative Estimation of BCR-ABL Transcript: An In-Lab Procedural Study on Leukemia PatientsОт EverandThe Qualitative Estimation of BCR-ABL Transcript: An In-Lab Procedural Study on Leukemia PatientsОценок пока нет
- Opeyemi IdaeworДокумент66 страницOpeyemi IdaeworOpeyemi IdaeworОценок пока нет
- Review of Related LiteratureДокумент11 страницReview of Related LiteratureJanОценок пока нет
- Hodgkin Lymphoma 2023 Update On Diagnosis, Risk-Stratification, and ManagementДокумент24 страницыHodgkin Lymphoma 2023 Update On Diagnosis, Risk-Stratification, and ManagementZuri100% (1)
- Chronic Lymphocytic Leukaemia: Key PointsДокумент5 страницChronic Lymphocytic Leukaemia: Key PointsJorge Tovar AvilaОценок пока нет
- Disorders of White Blood Cells and Lymphoid TissuesДокумент30 страницDisorders of White Blood Cells and Lymphoid Tissuesammar amerОценок пока нет
- Group 1 Apolonio, Leizel, R. Corpuz Rolyn Mañalac Joelle Anne Señeres Loui AnneДокумент33 страницыGroup 1 Apolonio, Leizel, R. Corpuz Rolyn Mañalac Joelle Anne Señeres Loui AnneLeizel ApolonioОценок пока нет
- LymphomasДокумент51 страницаLymphomasYousef IbrahimОценок пока нет
- Diseases and Tumors of Lymphoid SystemДокумент97 страницDiseases and Tumors of Lymphoid SystemamyОценок пока нет
- Thesis On Hodgkin LymphomaДокумент5 страницThesis On Hodgkin Lymphomafjm38xf3100% (2)
- An Sell 2005Документ11 страницAn Sell 2005Rachel AutranОценок пока нет
- Week 15-Mature-Lymphoid-Neoplasms-SCДокумент65 страницWeek 15-Mature-Lymphoid-Neoplasms-SCKyle CollladoОценок пока нет
- Hodgkin's DiseaseДокумент11 страницHodgkin's DiseaseReiswandhika Intan PermatasariОценок пока нет
- Acute Leukaemias Lecture-1Документ39 страницAcute Leukaemias Lecture-1MarvellousОценок пока нет
- Clinical Presentation, Pathologic Features, Diagnosis, and Differential Diagnosis of Chronic Lymphocytic Leukemia - UpToDateДокумент23 страницыClinical Presentation, Pathologic Features, Diagnosis, and Differential Diagnosis of Chronic Lymphocytic Leukemia - UpToDateJosé Martínez100% (1)
- LLC 2006Документ11 страницLLC 2006claudia8a_ulamedОценок пока нет
- Paediatric Acute Lymphoblastic LeukemiaДокумент49 страницPaediatric Acute Lymphoblastic LeukemiaKishoreChandraKoradaОценок пока нет
- Fast Facts: Blastic Plasmacytoid Dendritic Cell Neoplasm: Shedding light on a rare diseaseОт EverandFast Facts: Blastic Plasmacytoid Dendritic Cell Neoplasm: Shedding light on a rare diseaseОценок пока нет
- Reviews: Chronic Lymphocytic Leukaemia: From Genetics To TreatmentДокумент18 страницReviews: Chronic Lymphocytic Leukaemia: From Genetics To Treatmentlengers poworОценок пока нет
- Bosch2019 PDFДокумент18 страницBosch2019 PDFlengers poworОценок пока нет
- Leukaemia Lecture 01 - Aml - DRДокумент69 страницLeukaemia Lecture 01 - Aml - DRapi-273068056100% (1)
- Acute Lymphoblastic LeukemiaДокумент34 страницыAcute Lymphoblastic LeukemiamtyboyОценок пока нет
- K3. Penatalaksanaan Kasus Neurologi Di FKTP - Dr. Achmad Faqih, SP.SДокумент30 страницK3. Penatalaksanaan Kasus Neurologi Di FKTP - Dr. Achmad Faqih, SP.SLia pramitaОценок пока нет
- 388tetanus For Website PDFДокумент52 страницы388tetanus For Website PDFLia pramitaОценок пока нет
- Time Table-1-1Документ3 страницыTime Table-1-1Lia pramitaОценок пока нет
- Jadwal 1 Februari FixДокумент4 страницыJadwal 1 Februari FixLia pramitaОценок пока нет
- K28. Malpraktek - Dr. Monang, SP.FДокумент18 страницK28. Malpraktek - Dr. Monang, SP.FLia pramitaОценок пока нет
- Journal AppraisalДокумент1 страницаJournal AppraisalNur Vicka VickaОценок пока нет
- ESC Guidelines For DX and TX of Acute and Chronic HF (2016)Документ85 страницESC Guidelines For DX and TX of Acute and Chronic HF (2016)Imja94Оценок пока нет
- Ehz 467Документ65 страницEhz 467Coy Calapatia-TorresОценок пока нет
- K20. DNA Forensik - Dr. Virhan, M.BiomedДокумент20 страницK20. DNA Forensik - Dr. Virhan, M.BiomedLia pramitaОценок пока нет
- Prevention Guidelines Made SimpleДокумент17 страницPrevention Guidelines Made SimplearseniosilvaОценок пока нет
- Lung Abscess: DR Budi Enoch SPPDДокумент14 страницLung Abscess: DR Budi Enoch SPPDLia pramitaОценок пока нет
- Systrmic Lupus Erythematosus (Sle)Документ62 страницыSystrmic Lupus Erythematosus (Sle)Dedy Santoso ChouОценок пока нет
- Steno Ciii StenoДокумент21 страницаSteno Ciii StenoLia pramitaОценок пока нет
- Atherosklerosis: DR Budi Enoch SPPDДокумент56 страницAtherosklerosis: DR Budi Enoch SPPDLia pramitaОценок пока нет
- Myeloid LeukemiaДокумент36 страницMyeloid LeukemiaLia pramitaОценок пока нет
- ReferatДокумент53 страницыReferatLia pramitaОценок пока нет
- Gout and Othercrystal-Associated ArthropathiesДокумент31 страницаGout and Othercrystal-Associated ArthropathiesLia pramitaОценок пока нет
- Systemic Lupus Erythematosus (SLE)Документ62 страницыSystemic Lupus Erythematosus (SLE)Lia pramitaОценок пока нет
- Atherosklerosis: DR Budi Enoch SPPDДокумент56 страницAtherosklerosis: DR Budi Enoch SPPDLia pramitaОценок пока нет
- Coagulation DisordersДокумент26 страницCoagulation DisordersLia pramita0% (1)
- Hemoglobinopathy Dan Sindroma ThalasemiaДокумент62 страницыHemoglobinopathy Dan Sindroma ThalasemiaLia pramitaОценок пока нет
- Coagulation DisordersДокумент26 страницCoagulation DisordersLia pramitaОценок пока нет
- Periarticular Disorders of The ExtremitiesДокумент20 страницPeriarticular Disorders of The ExtremitiesLia pramitaОценок пока нет
- Gout and Othercrystal-Associated ArthropathiesДокумент31 страницаGout and Othercrystal-Associated ArthropathiesLia pramitaОценок пока нет
- 5 Pain Management in The Elderly PopulationДокумент26 страниц5 Pain Management in The Elderly PopulationLia pramitaОценок пока нет
- Gout and Othercrystal-Associated ArthropathiesДокумент31 страницаGout and Othercrystal-Associated ArthropathiesLia pramitaОценок пока нет
- 4 Palliative CareДокумент35 страниц4 Palliative CareLia pramitaОценок пока нет
- Glucose and Dementia - Geriatri JurnalДокумент20 страницGlucose and Dementia - Geriatri JurnalLia pramitaОценок пока нет
- Geriatric Medicine KuliahДокумент38 страницGeriatric Medicine KuliahLia pramitaОценок пока нет
- AsthmaICS PDFДокумент7 страницAsthmaICS PDFCARES Martin Federic PalcoОценок пока нет
- Procedures in OBGДокумент8 страницProcedures in OBGKanak Soni100% (1)
- Modul Praktikum Patologi Anatomi Fakultas Kedokteran Universitas Muhammadiyah Palembang Tahun 2015Документ23 страницыModul Praktikum Patologi Anatomi Fakultas Kedokteran Universitas Muhammadiyah Palembang Tahun 2015TasyaОценок пока нет
- DataДокумент207 страницDatamikhael muaneoОценок пока нет
- Assessment, Traumatic Grief, TahomaДокумент3 страницыAssessment, Traumatic Grief, TahomaJesus Carrillo Raymond100% (2)
- Sample Questions 2Документ6 страницSample Questions 2Filipino Nurses CentralОценок пока нет
- Bowel Nosode MatrixДокумент3 страницыBowel Nosode MatrixAndrea Middleton92% (13)
- Hiking and TrekkingДокумент11 страницHiking and TrekkingDavidОценок пока нет
- 5 - Cell Division - Notes 1Документ68 страниц5 - Cell Division - Notes 1Cole HarrisОценок пока нет
- Abstracts 8th Congress CSMBLM Rijeka Korekcija N.nikolac 5.11.2015. BO2Документ101 страницаAbstracts 8th Congress CSMBLM Rijeka Korekcija N.nikolac 5.11.2015. BO2BoyITBОценок пока нет
- Benign Prostatic Hyperplasia: Pembimbing: DR Herinto Himawan Sp.UДокумент42 страницыBenign Prostatic Hyperplasia: Pembimbing: DR Herinto Himawan Sp.Uilma aulia zahraОценок пока нет
- Dentassure Gano Toothpaste Launch PDFДокумент14 страницDentassure Gano Toothpaste Launch PDFnitesh sharma100% (1)
- Surgical Endoscopy Journal 1Документ86 страницSurgical Endoscopy Journal 1Saibo BoldsaikhanОценок пока нет
- Worry More Live Longer American English Student Ver2 BWДокумент5 страницWorry More Live Longer American English Student Ver2 BWLanting LeeОценок пока нет
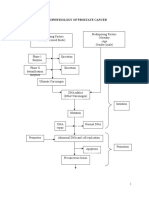
- Pathophysiology of Prostate CancerДокумент3 страницыPathophysiology of Prostate Cancermkho100% (1)
- Nej Mo A 1905287Документ12 страницNej Mo A 1905287Ari KurniawanОценок пока нет
- Opioid-Induced Androgen Deficiency in Males (OPIAD Syndrome) : Medicinski Casopis January 2015Документ7 страницOpioid-Induced Androgen Deficiency in Males (OPIAD Syndrome) : Medicinski Casopis January 2015Lilamala LilavelikaОценок пока нет
- Periodontal Risk Assessment, Diagnosis and Treatment PlanningДокумент22 страницыPeriodontal Risk Assessment, Diagnosis and Treatment PlanningHenry E. CoaquiraОценок пока нет
- KalojiraДокумент2 страницыKalojiralutfurrahman35Оценок пока нет
- Material Safety Data Sheet: 1. Chemical Product and Company IdentificationДокумент7 страницMaterial Safety Data Sheet: 1. Chemical Product and Company IdentificationKun Adi ReksatamaОценок пока нет
- PMP Pancreas 2014Документ5 страницPMP Pancreas 2014Lyna PangaribuanОценок пока нет
- Chapter 6 - Inflammation, Tissue Repair & FeverДокумент8 страницChapter 6 - Inflammation, Tissue Repair & FeverChrista CastorОценок пока нет
- GuidelinesДокумент4 страницыGuidelinesChidambaram SeevakanОценок пока нет
- HDFC Life Click 2 Protect Life - Product Brochure PDFДокумент24 страницыHDFC Life Click 2 Protect Life - Product Brochure PDFvaibhav kumar KhokharОценок пока нет
- BIOGENДокумент71 страницаBIOGENPamela GabrielОценок пока нет
- UPTODATE - MIOMA - Epidemiology, Clinical Manifestations, Diagnosis, and Natural History of Uterine Leiomyomas (Fibroids)Документ20 страницUPTODATE - MIOMA - Epidemiology, Clinical Manifestations, Diagnosis, and Natural History of Uterine Leiomyomas (Fibroids)Gaby FloresОценок пока нет
- Functions of The Respiratory PassagewaysДокумент2 страницыFunctions of The Respiratory PassagewaysAdnin ShereenОценок пока нет
- Functional Dyspepsia: Recent Advances in Pathophysiology: Update ArticleДокумент5 страницFunctional Dyspepsia: Recent Advances in Pathophysiology: Update ArticleDen BollongОценок пока нет
- Building An NCD-ready Workforce: Technical Meeting ReportДокумент48 страницBuilding An NCD-ready Workforce: Technical Meeting ReportnavenОценок пока нет
- GYNE 9 1 Breast Diseases PDFДокумент19 страницGYNE 9 1 Breast Diseases PDFColeen NeyraОценок пока нет